Reflections on SITC (Part 2)
Part 2: Biomarkers and Tumor Biology
Bill Slichenmyer, M.D., Sc.M
Alacrita Consulting (@alacrita.com)
The Society for Immunotherapy of Cancer (SITC) convened its 31st annual meeting in early November. Shortly after the meeting I posted some thoughts on data presented there regarding new drugs in development. In this post my goal is to continue with more data from the SITC meeting, this time with a focus on biomarker data that piqued my interest. In addition, this post also includes comments on two important papers that were published during the week of the SITC meeting. A few references are cited at the end of the post.
Comparing PD-L1 Assays: They’re Not Identical
The PD-1 receptor is a key molecular target in oncology therapeutics. Expression of one of its ligands, PD-L1, is routinely assayed in tumor tissue using immunohistochemistry (IHC). PD-L1 expression has emerged as an important predictive biomarker for selection of patients with anti-PD-1 therapy in clinical practice, especially in 1st line non-small cell lung cancer (NSCLC).
Clinical trial results have been reported in the literature for PD-L1 IHC assays using at least seven different anti-PD-L1 antibodies. Three different assays have been approved by the FDA for various indications (using antibodies 22C3 and 28-8 from Dako as well as SP142 from Ventana). The IHC assays vary from one another due to potential differences in: 1) the antibody’s binding site on PD-L1, 2) binding affinities, 3) inclusion of tumor cells and/or inflammatory cells in scoring and 4) cutoff levels for the percent positive cells in a sample. This this has led to difficulty in comparing results across clinical trials that used different assays.
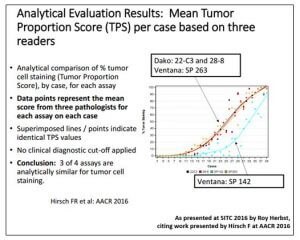
In a SITC session on State-of-the-Art Immunotherapies, Roy Herbst (Yale) gave a presentation on what’s new in NSCLC [1]. One of the topics he addressed is the ongoing industrial/academic collaborative effort known as the Blueprint Project, which is comparing PD-L1 IHC diagnostics for immune checkpoint inhibitors. Dr. Herbst showed results (previously presented at AACR 2016 by Fred Hirsch and subsequently published [2]) that compared the percent of tumor cell staining for four anti-PD-L1 antibodies in 39 NSCLC samples.
As shown in the nearby figure, three of the four antibodies are analytically similar while one of them (SP142) appears to be less sensitive than the others. The implication is that a direct comparison across studies (which is always challenging) will be even harder when one of the trials used a PD-L1 assay based on the SP142 antibody. For those planning future clinical trials, it will be important to consider these results when selecting a PD-L1 assay for defining eligibility criteria, stratification factors and/or correlative analyses. As discussed below, multiplexed assays of various immune biomarkers can provide additional information that is complementary to PD-L1 status.
Mutational Load and the Inflamed Tumor Microenvironment (TME):
They’re not correlated!
It has been previously shown that response to checkpoint inhibitors correlates with intratumoral mutational load [3] and with an inflamed TME [4]. A keynote speaker at the SITC meeting, Ira Mellman (Genentech), mentioned that he and colleagues will soon publish data showing that mutational load is constant across inflamed and non-inflamed tumors [5]. This point was demonstrated in a SITC poster by authors from Merck, Fox Chase and Dana-Farber [6]. These authors initially analyzed data on mutational load and an “IFN-g centric” gene expression profile of TME inflammation from patients in two trials of pembrolizumab (Keytruda, Merck) in patients with solid tumors. They confirmed their findings in independent databases from Moffitt and TCGA. They concluded that mutational load and TME inflammation across tumor types are independent and complementary biomarkers of response to pembrolizumab.
On November 11, the very day of Dr. Mellman’s SITC lecture, two relevant papers on this topic were published online in PNAS: one from authors at the University of Chicago [7, Spranger et al.] and another from Johns Hopkins [8, Danilova et al.]. The Danilova study examined PD-1, PD-L1 and PD-L2 expression, mutational density and gene signatures for cytolytic activity and Th1/ IFN‑g activity in ~3,500 patients across nine different solid tumor types in the TCGA database. The paper describes findings in certain tumor types, such as the observation in RCC and squamous NSCLC that PD-L1 expression is not associated with T-cell cytolytic activity or the IFN-g gene signature. Their data suggest that the combination of PD-L1 and IFN-g gene signature would be a better predictor of anti-PD-1 response than the PD-L1 biomarker alone. Across various tumor types they found no direct association between mutational load and PD-L1 expression, T-cell cytolytic activity or the IFN-g gene signature. They conclude that their findings support the use of multiplexed biomarker analyses that are tumor-type specific and incorporate both expression and genomic profiling.
The work described in the Spranger paper initially focused on 266 melanoma cases in the TCGA database. They showed that inflamed and non-inflamed melanoma samples contain comparable numbers of neoantigens. They studied these neoantigens in vitro and concluded that the inflamed vs. non-inflamed samples did not differ in their ability to bind to MHC I. The non-inflamed samples showed reduced expression of genes associated with CD141+ Batf3-lineage dendritic cells. They went on to expand their analysis of mutation density vs. TME inflammation across multiple tumor types in the TCGA database.
Below is a figure from the Spranger paper showing the relationship across multiple tumor types between mutational load (y-axis) and inflammation status. The latter is depicted by the color of each dot, which represents one patient’s tumor: red (inflamed), gray (intermediate), blue (non-inflamed).
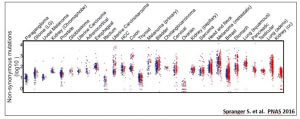
The authors discuss factors that may explain why certain tumors with a high mutational load may lack a T-cell inflamed TME, such as failed recruitment of Batf3-lineage dendritic cells. They conclude the paper on a hopeful note, observing that noninflamed tumors express sufficient antigens for potential recognition by CD8+ cells. They look forward to the development of novel therapies to restore T-cell activation and trafficking with the aim of restoring responsiveness to checkpoint blockade. An example of a drug that is being explored in this manner is described below.
A Mek Inhibitor May Have a Role in Immunotherapy
Interesting biomarker data were presented during the session on New Agents in Development by Edward Cha (Genentech) [9]. He discussed the development of the combination of the anti-PD-L1 checkpoint inhibitor atezolizumab (Tecentriq, Genentech/Roche) in combination with the mek inhibitor cobimetinib (Cotellic, Genentech/Roche). He cited previously published work that mek inhibition increases MHC I expression and the number of antigen-specific CD8+ T cells in tumor bearing mice [10].
The combination of atezolizumab with cobimetinib is being explored in the clinic. Preliminary results of a phase 1b study of the combination were presented by Johanna Bendell at ASCO 2016 [11]. At SITC Dr. Cha reviewed some of the biomarker data from that presentation. Below is a figure showing an impressive, albeit anecdotal, comparison of archival pre-treatment tumor tissue vs. effects following dosing with cobimetinib and then again after the combination of cobimetinib with atezolizumab. In this patient with a clear cell sarcoma it appears that cobimetinib has increased CD8+ infiltration and MHC I expression in the TME as had been reported in the mouse model. Biomarker data such as these can be useful to guide drug development.
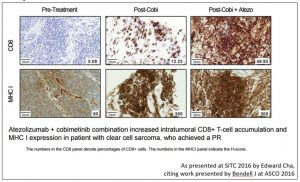
At ASCO, Dr. Bendell mentioned that encouraging clinical responses had been seen in patients with microsatellite stable colorectal cancer, a population generally considered to be unresponsive to checkpoint inhibitor therapy. The cobimetinib + atezolizumab combination is currently being studied in a company- sponsored randomized controlled trial in patients with 3rd line colorectal cancer [12]. The study has a planned sample size of 360 patients. The primary endpoint, overall survival, is expected to read out in 2019. The results will be of considerable interest.
The SITC meeting is a great setting to see and hear about new developments in immuno-oncology. I hope to see you there next year!
December 29, 2016
REFERENCES
1. Herbst R. Lung Cancer: Frontline Therapy, Predictive Markers, and Novel Combinations. SITC Annual Meeting, 2016.
2. Hirsch FR et al. PD-L1 Immunohistochemistry Assays for Lung Cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J Thorac Oncol. 2016 Nov 29 [Epub ahead of print].
3. Snyder A et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. NEJM 371:2189-99, 2014.
4. Tumeh PC et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568-571, 2014.
5. Mellman I. The Mechanistic Basis of Cancer Immunotherapy. SITC Annual Meeting, 2016.
6. Cristescu R et al. Tumor mutational load and T cell inflamed microenvironment are independent determinants of response to pembrolizumab. Abstract P72, J Immunother Cancer 4: (Supp 1), 2016
7.Spranger S et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. PNAS 113: E7759–E7768, 2016.
8. Danilova L et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. PNAS 113: E7769–E7777, 2016.
9. Cha E. Cobimetinib in Combination with Atezolizumab. SITC Annual Meeting, 2016.
10. Ebert PJ et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 44:609-21, 2016.
11. Bendell J et al. Clinical activity and safety of cobimetinib and atezolizumab in colorectal cancer (CRC). J Clin Oncol 34, 2016 (suppl; abstr 3502).
12. NCT02788279 on ClinicalTrials.gov: https://clinicaltrials.gov/ct2/show/NCT02788279?term=NCT02788279&rank=1