The cell therapy field has been predominantly active in the oncology space and is enjoying bench-to-bedside clinical success. However, the cancer cell therapy space has become very crowded and drug developers are currently seeking to expand the field into autoimmunity, with biotech and pharmaceutical companies employing a variety of strategies to develop differentiated cellular therapies against autoimmune diseases that present a huge unmet need.
It is canonically fundamental that the immune system recognizes and responds to foreign pathogens, whilst remaining unresponsive to host tissues and self-antigens 1. This immunological tolerance is based on regulatory mechanisms that counterbalance the effector immune system. In autoimmune diseases, this intricate balance is lost, resulting in cell and tissue destruction by self-reactive cytotoxic T cells or autoantibodies, and the accompanying inflammatory process 2.
There are over 80 known autoimmune diseases, the most common being psoriasis, type I diabetes (T1DM), and rheumatoid arthritis. Autoimmune diseases can be systemic such as systemic lupus erythematosus (SLE), or organ specific such as T1DM which mostly affects the pancreas. Over 4% of the world’s population suffers from one or more of these diseases 3.
Existing therapies for autoimmune diseases
The majority of autoimmune diseases are chronic and there is no definitive cure. The complexity and heterogeneity of autoimmune diseases has greatly impeded the development of efficacious targeted treatments, as they are required to sufficiently purge the immune system of autoreactivity while maintaining its functional side 4.
Earlier treatment of autoimmune disorders focused on managing end-organ manifestations such as insulin replacement in diabetes and alleviating pain and inflammation in arthritis. The current standard of care includes non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids and disease-modifying anti-rheumatic drugs (DMARDs). NSAIDs and glucocorticoids reduce inflammation and alleviate pain, while DMARDs are effective in reducing the damaging effect of the inflammatory autoimmune response 5.
Newer, more targeted therapies aim to neutralize key proinflammatory cytokines, interfere with and inhibit the activation of lymphocytes, or enhance the pathways that suppress these cells. For example, abatacept (Orencia, Bristol Myers Squibb), a chimeric fusion protein CTLA-4-IgG1 is used for the treatment of rheumatoid arthritis and psoriasis, and acts as a co-stimulatory blockade that inhibits autoreactive T cell activation 6. TNF inhibitors are the most widely used biologic therapy. Anti-TNF biologics such as Humira (AbbVie), Remicade (Pfizer), Enbrel (Pfizer) and Simponi (Janssen Biotech), have improved the management of autoimmune disorders 7. However, a significant proportion of patients fail to respond to treatment. For example, a common mechanism implicated in loss of response is immunogenicity due to the formation of antibodies against TNFα antagonists 8. The reasons for failure of immunosuppressive are often attributed to the metabolic or pharmacokinetic property of the drugs 9.
Even for those that do respond to treatment, drug-free remission or cure is rare, and most patients require life-long maintenance therapy. Over one third of rheumatoid arthritis patients fail to respond to conventional therapies, and 50% of those that do respond experience flare-ups if treatment is discontinued 10. These numbers are mirrored in other autoimmune diseases such as SLE and Crohn’s disease 11, 12. Furthermore, immunosuppressants are associated with side effects such as nausea and vomiting and long-term use leaves patients susceptible to opportunistic infections and malignancies.
There has been a push for the development of long-term therapies that re-establish stable immune tolerance, have low toxicity with few side effects, and reduce the burden of needing continuous medication. Importantly, there is a pressing need for therapeutic drugs that specifically target pathogenic cells whilst leaving the remainder of the immune system functioning normally.
Tolerogenic cell therapy
Recent drugs in development are focusing on promoting the induction and expansion of cells known to confer immune tolerance. The mTOR inhibitor rapamycin, IL-10, low dose IL-2, tumour necrosis factor receptor 2 (TNFR2) agonists or FMS-like tyrosine kinase 3 ligand (FLT3L), have been explored. The mTOR inhibitor rapamycin has the ability to promote Treg cell proliferation and stimulate TCR-induced T cell anergy and deletion. This effect is enhanced with the administration of IL-10 13, or FLT3L through selective expansion of plasmacytoid dendritic cells (DCs) 14. Low-dose IL-2 administration is effective by selectively targeting the high CD25-expressing Tregs 15. TNFR2 elicits strong anti-inflammatory effects and was shown to kill autoreactive CD8+ cells in T1DM 16. Administration of disease-specific autoantigens or autoantigenic peptides as a vaccine against autoimmunity has been investigated to desensitize the autoimmune system and activate antigen-specific Tregs. In mouse models, tolerance to these antigens has been demonstrated across various autoimmune diseases 17. However, due to the complexity of immune tolerance mechanisms that are not yet fully understood, translation to the clinic has been slow. The focus has therefore recently shifted to the administration of tolerogenic cells themselves. This has been made possible alongside advancements in cellular expansion and differentiation, and efficient manufacture of cellular therapies 18. There are six main types of tolerogenic cell therapies currently in development.
Mesenchymal stem/stromal cells (MSCs)
MSCs are adult stem cells with multipotent differentiation capacity. Pre-clinical studies suggest they have strong anti-inflammatory properties as well as the ability to promote regeneration. MSCs have the ability to skew the tolerogenic profiles of effector cells such as suppressing CD4+ and CD8+ T lymphocytes and promoting their differentiation into regulatory T cells (Tregs); inhibiting differentiation of B-cells into plasma cells and directly impeding B-cell proliferation, differentiation and antibody production; as well as the activation and expansion of natural killer (NK) cells 19. They carry out these activities by directly interacting with immune effector cells or through the induction of specific factors, such as indoleamine 2,3-dioxygenase, prostaglandin E2, transforming growth factor β (TGF-β), and IL-10, among others 20. However, their action and properties strongly depend on the inflammatory milieu and disease setting.
In addition to their regenerative and immunomodulatory properties, MSCs have the advantage of displaying low immunogenicity as they lack Class II MHC expression and costimulatory molecule expression such as CD40, CD80 and CD86, with an inability to stimulate T cell activation 21. Their unique immunological profile allows for allogeneic transfer with minimal risk of rejection and presents the possibility of a cell bank system for widespread use from a limited number of donors.
However, even though MSCs were identified 50 years ago and have since been explored for the potential treatment of autoimmune disorders since the early 2000s, their translation to the clinic has been disappointing. One of the main challenges surrounding MSCs is their heterogeneity. The quality of the MSC product is largely dependent on isolation and culture methods, as well as the age and medical history of the donor 22. Isolating an effective population of MSCs from patients with certain inflammatory diseases, such as rheumatoid arthritis or diabetes has proven challenging and researchers suggest possible loss of therapeutic function by these cells 23. Recent studies have furthermore disputed their ‘immune privilege’ as in vivo observations suggest that mismatched allogeneic MSCs do not persist in patients following infusion, and were shown to induce a humoral and cellular immune response in host mice 24, 25. The popularity of MSCs has thus waned.
Regulatory T (Treg) cells
Treg cells play a central role in the maintenance of immune tolerance and are thus critical in the prevention of autoimmune diseases. They are a subset of CD4+ T cells expressing CD25 and transcription factor Forkhead Box P3 (FoxP3) that impede effector T cell function 17. Treg cells can confer immune tolerance through the expression of anti-inflammatory mediators or cell-to-cell contact. Importantly, they can also act in an antigen non-specific manner through bystander suppression, whereby Treg cells propagate their suppressive properties to neighboring cells 26. This unique ability of Treg cells allows for targeting of inflamed and damaged tissue. Treg defects, in terms of decreased cell number and suppressive phenotype are known to be associated with autoimmune disorders 17. Therefore, in order to restore immune tolerance, the administration of Treg cells has the potential to treat autoimmune disease and has thus emerged as a focus for cell-based therapies in the past decade. Researchers have considered a wide variety of approaches to enhance Treg cell number, specificity and function. Current approaches include administration of non-specific polyclonal Tregs, or Tregs that have been engineered to express a T cell receptor (TCR) or a chimeric antigen receptor (CAR) specific for the autoantigen inducing the unwanted immune response.
Polyclonal Treg cells
Polyconal Tregs express a natural repertoire of TCRs. Autologous polyclonal Treg cells, isolated from peripheral blood and expanded in vitro are the most widely clinically tested Treg cell therapy. Administration of polyclonal Tregs has been shown to be safe in T1DM 27, as well as in other autoimmune diseases and organ transplant patients. However, these cells did not survive long-term in patients and have failed to provide a meaningful clinical benefit.
It is thought this may be because Tregs are non-antigen specific, as the natural repertoire of TCRs target a broad mix of antigens 28. It is also believed that the unmodified T cells were not potent enough and difficult to isolate and expand due to low precursor frequencies. Drug developers are optimising Treg therapy by engineering the cells to be autoantigen-specific. Numerous preclinical studies have demonstrated that antigen-specific Tregs are more potent than unselected polyclonal Tregs, making them better equipped to restore immune tolerance, with a higher likelihood of complete remission 29, 30.
There are also some concerns surrounding polyclonal Treg encouragement of generalised immune suppression, increasing the risk of infection and cancer. However, antigen-specific Treg cells may demonstrate greater safety as they predominantly localise at the site of antigen presentation, decreasing the risk of general immunosuppression 26. Researchers are exploring several approaches to present antigen specificity to Tregs and expand them in vitro. One approach involves redirecting polyclonal Tregs by introducing synthetic receptors. The next generation of Treg cell therapy will see cells engineered ex-vivo with CAR or TCR constructs, as well as modifications that enhance persistence, potency and stability.
TCR-Treg cells
The genetic engineering of a TCR on Tregs allows autoantigens to be targeted in the context of an antigen-MHC-peptide complex. Current TCR-Tregs in development are ex-vivo transduced to express a high affinity, autoantigen-specific TCR. The first-in-human study using TCR-Treg therapy was the transfer of ovalbumin (OVA)-specific TR1 cells against Crohn’s disease 31. The triggering antibody for inflammatory bowel disease is not known, but food antigen ovalbumin and its distribution along the digestive tract has proven to be effective in activating Tregs locally 32. The treatment was shown to be safe and effective. The field has since broadened, exploring the potential of TCR-Tregs in a variety of autoimmune diseases.
Current efforts have involved antigen-specific TCRs isolated from effector T cells and transduced into Treg cells. However, it is possible that TCRs isolated from Treg cells have a greater intrinsic affinity and specificity, affecting migration and functional activity 26. Thus, an approach isolating TCRs from Tregs may more closely replicate activity of natural Treg cells. Although the number of antigen-specific Tregs required is significantly less than polyclonal Tregs, the identification of an appropriate, high-affinity, autoantigen-specific TCR presents a challenge for some autoimmune diseases with poorly defined dominant epitopes. The great diversity in TCRs, coupled with the fact that few antigen-specific Tregs are naturally circulating in the peripheral blood, makes them hard to isolate and identify 17.
CAR-Treg cells
CARs are also being expressed in Tregs as a strategy to improve the potency and specificity of Treg therapies by recognising epitopes on surface-expressed molecules. The main advantage of CARs is the ability to identify the target antigen in an MHC independent manner, thus overcoming limitations posed on TCR-Tregs 33. This also bypasses the requirement for matching of the patient MHC genotype, making it suitable for a larger population of patients 26. However, they have the disadvantage of being unable to recognise intracellular targets. Additionally, they need approximately 100 targeted autoantigens on a cell surface for the CAR to recognise it and stimulate the Treg, contrasted with the requirement of potentially only one peptide-MHC interaction with TCR in order for the Treg to become activated 17. Given the advantages and disadvantages of TCR- and CAR-Tregs, further research is required to provide insight into which technology is more appropriate for a particular context.
A few drug developers are also exploring effector CAR-T therapy for autoimmune diseases. However, CAR-Tregs are likely to display greater persistence and durable immunosuppression in the target tissue 34. Furthermore, effector CAR-T cell therapies could result in substantial tissue damage. Off-target effects of CAR-Tregs are expected to be less severe, potentially including prolonged immunosuppression at non-diseased tissues, opportunistic infections, or suppression of local tumor immunity.
CAR-NK cells
Engineering NK cells to express CARs is believed to yield greater safety and clinical feasibility in cancer 35. This is yet to be explored in autoimmunity and the biological rationale is not clear.
Tolerogenic dendritic cells (tolDCs)
DCs play an important role in the balance between immunity and tolerance. In autoimmune diseases, DCs tend to skew to a mature proinflammatory state, secreting proinflammatory cytokines and activating autoreactive T cells 18. tolDCs are a subset of DCs able to prime the immune system in a tolerogenic state. They exhibit an immature phenotype, characterised by lower expressions of CD40, CD80, CD86, MHCII, and the production of anti-inflammatory cytokines, such as high levels of IL-10 and TGF-β and low levels of IL-2 36. Their activity promotes the inhibition of autoreactive T cells, as well as the activation and proliferation of Tregs. tolDC cell therapy offers an opportunity to expand antigen-specific Treg cells, enhancing the antigen-specific suppressive properties of Tregs 37.
The Landscape
Technology type
There are currently two launched cell therapy products for autoimmune diseases, both allogeneic MSCs. Mesoblast and JCR Pharmaceuticals launched remestemcel-L in 2016 in Japan for children and adults with acute graft versus host disease (GvHD) following hematopoietic stem cell transplantation (HSCT). It is also in clinical development for GvHD and Crohn’s disease in the US. Takeda launched Alofisel in Europe in 2018, the first allogeneic stem cell therapy to receive central marketing authorization approval in Europe 38. It is indicated for refractory complex perianal fistulas in Crohn's disease. The therapy comes with a moderately high price tag, with NICE noting its list price at £54,000 39.
The current global development pipeline consists of over 50 cell therapies to treat autoimmune disorders. Over half of these are accounted for by MSCs (Figure 1), the majority of which are in late clinical development. The next most advanced stage cell therapy is polyclonal Treg cell therapy (Figure 2). More recently, antigen-specific engineered Tregs and CAR-Tregs have rapidly emerged, all of which are pre-clinical stage. CAR-T and CAR-Tregs are currently the second most common type of cell therapy in development as they offer highly targeted and stable suppressive effects. Drug developers are leveraging the orthogonal benefits from CAR-T cells in the oncology space, capitalizing on new tools for boosting potency and persistence. For example, researchers are assessing the addition of co-stimulatory molecules, a procedure that has greatly optimized efficacy in the oncology space. Most CAR Treg studies to date have used an identical design to 2nd generation CARs in cancer that includes a single co-stimulatory domain linked to the primary CD3ζ signaling domain 33. There is also one DC program and one CAR-NK program in development.
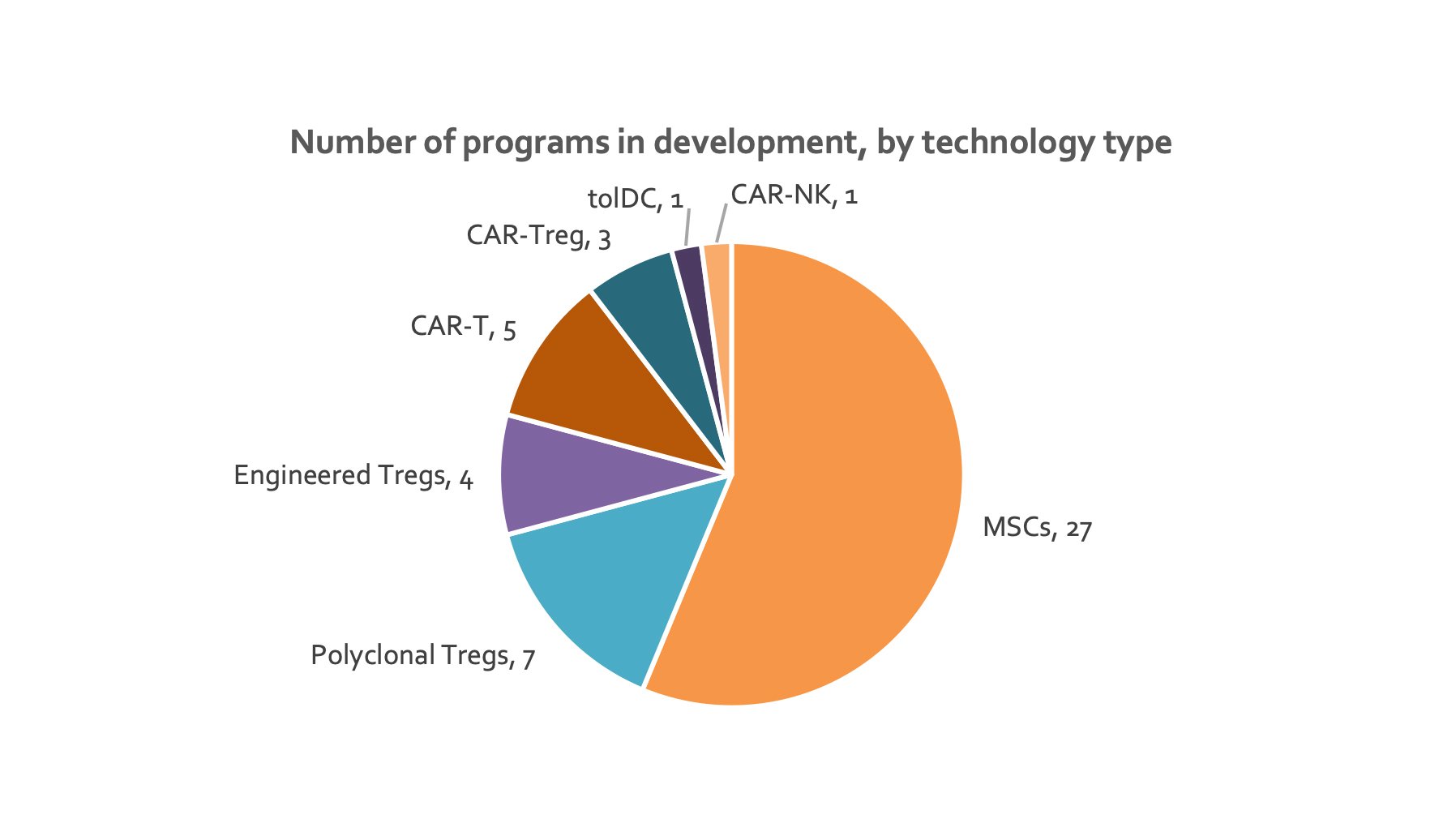
Figure 1. Pie chart depicting number of therapies for each type of cell therapy (data from individual company websites).
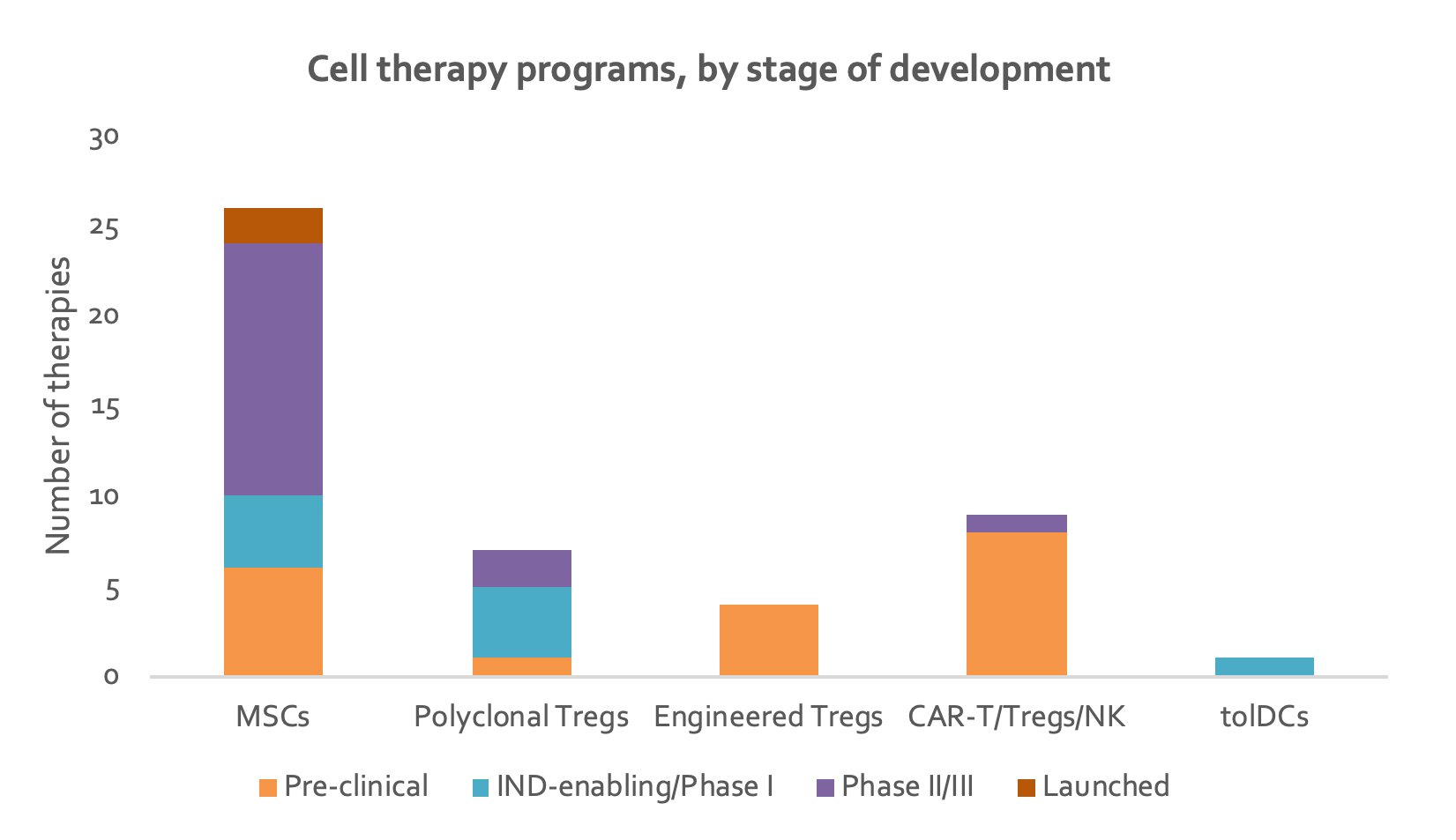
Figure 2. Stacked column chart depicting number of therapies in each stage of development for each type of cell therapy (data from individual company websites).
MSCs
Mesoblast is the only company with more than one cell therapy product against autoimmune disorders. In addition to remestemcel-L, Mesoblast is developing MPC-300-IV, an allogeneic mesenchymal precursor cell therapy for the treatment of diabetes and rheumatoid arthritis. Mesoblast's proprietary mesenchymal lineage adult stem cells technology platform develops the earliest precursors of the mesenchymal cell lineage in adult tissues, which has the benefit of displaying lower immunogenicity 40.
Imstem Biotechnology is developing a MSC therapy derived from embryonic stem cells. Their proprietary method to derive MSCs from embryonic stem cells via a trophoblast-like intermediate stage outperforms other methods by 200 times in cell yield, 80% shorter processing time, and 75% lower manufacturing cost 41. Additionally, embryonic stem cells display significant advantages over traditional adult-tissue derived MSCs such as ease of availability and no batch, donor or manufacture variance 42. They are also resistant to changes in the environment after expansion and infusion and have low immunogenicity. Similarly, Citius pharmaceuticals is developing a MSC therapy derived from mRNA-reprogrammed induced pluripotent stem cells, using Novellus and Factor Bioscience’s technology. The manufacturing process facilitates the robust expansion of uniform MSCs that have higher potency, secrete higher levels of immunomodulatory proteins and offer practically unlimited supply 43.
SCM Lifescience is unique as it uses its proprietary stem cell isolation technology to develop high purity allogeneic MSCs and stores them in a cell bank system 44. Orbsen Therapeutics has also developed an approach to enable the isolation of a clearly defined cellular population. The company identified a marker protein as a tool for selecting 100% pure therapeutic stromal cells from a mixture of cells residing in tissues. Their proprietary technology is more precise at isolating and harvesting stromal cells than the current methods employed by most stem cell developers who rely on cell adherence to plastic cell culture dishes 45. Orbsen Therapeutics has achieved the highest levels of MSC purity yet 46.
Polyclonal Treg cells
Organisations developing polyclonal Treg cell programs include King's College London, University of California San Francisco, Cellenkos, Tract Therapeutics and Regenex.
Acer Therapeutics is developing Tcelna, a vaccine consisting of radiation-attenuated autologous T cells for the treatment of multiple sclerosis. Tcelna is manufactured using Opexa's proprietary method, specifically isolating myelin-reactive T cells and raising them against selected peptides. Opexa evaluates over 100 peptides across the three key myelin proteins to identify each patient's set of dominant epitopes 47. Atara Bio and QIMR Berghofer are developing Epstein–Barr virus-specific cytotoxic T-lymphocytes, for the treatment of multiple sclerosis.
Genetically engineered Treg cells
Casebia has combined its CRISPR-Cas9 gene editing technology with Seattle Children's Hospital’s T cell manufacturing technology to develop CRISPR/Cas9 gene-edited Treg cells. GentiBio is using its proprietary platform to generate EngTreg cell therapy products that are antigen specific and tuneable, and selectively restrict inflammation temporally and spatially in specific tissues where it is beneficial. Rather than isolating Tregs, a rare cell population in the blood, GentiBio focuses on converting more abundant effector T cells into EngTreg cells 48. Sonoma Biotherapeutics and Lyell Immunopharma have collaborated to develop genetically engineered Treg cells.
CAR-T cells
There are currently eight CAR-T cell therapy programs against autoimmune diseases in development. Out of those, three are CAR-Treg cell therapies. Most of the organisations have yet to disclose their lead targets and indications.
Descartes-08 (Cartesian Therapeutics/Harvard Medical School) is the most advanced CAR-T program, in clinical development 49. Cartesian Therapeutics is developing CD8+ CAR-T cells by mRNA transfection, which target BCMA in myasthenia gravis 50 and claim to be harnessing “enhanced safety features” that presumably provide an advantage over the use of anti-BCMA mAb therapy. CRISPR Therapeutics have partnered with ViaCyte to evade rejection of Viacyte’s PEC-Direct in diabetic patients. CRISPR Therapeutics is using CRISPR-Cas9 technology to develop allogeneic CAR-Ts recognizing an undisclosed target with the aim of protecting transplanted cells 51. Atara Biotherapeutics is developing a CAR-T therapy using its novel co-stimulation technology designed to improve persistence and safety 52. China Immunotech is employing its proprietary antigen receptor complex structure which improves survival time of T cells in vivo and results in reduced T cell depletion 53.
Cabaletta Bio and University of Pennsylvania are developing chimeric autoantibody receptor T-cell, or CAAR-T therapies, for autoimmune diseases. CAARs are similar to typical CARs, with the exception that the extracellular domain carries antigen fragments instead of antibody fragments, targeting self-reactive B cells that cause autoimmune diseases and hence flip the original antibody-antigen interaction 54. A single CAAR T treatment could offer complete and durable remission of certain specific B cell-mediated autoimmune diseases while leaving the protective antibody-producing immune system intact. The company’s initial focus will be on mucosal pemphigus vulgaris 55. Cabaletta Bio is currently developing CAAR-Ts targeting muscle-specific tyrosine kinase (MuSK) against MuSK in myasthenia gravis 56.
Sangamo Therapeutics, Quell Therapeutics, and Kyvrena/Gilead are developing CAR-Treg cell programs. Gilead entered a partnership with Kyverna, combining Kyverna’s synthetic Treg platform with the synthetic gene expression system synNotch from Gilead’s Kite unit 57.
CAR-NK cells
Cincinnati Children's Hospital Medical Center is developing an anti-PD-1 CAR-NK cell therapy for the treatment of lupus. This will selectively target T follicular helper (Tfh) cells. A CAR with relatively weak binding to PD-1 was engineered, in order to eliminate Tfh cells that exhibit high expression of PD-1, while cells with lower levels of PD-1, including Treg and memory T cells are spared 35.
Indications
The majority of cell therapy programs are being developed for the treatment of Crohn’s disease, multiple sclerosis and rheumatoid arthritis (Figure 3). These are mostly MSC therapies. Lead indications for CAR-Treg programs have not yet been disclosed.
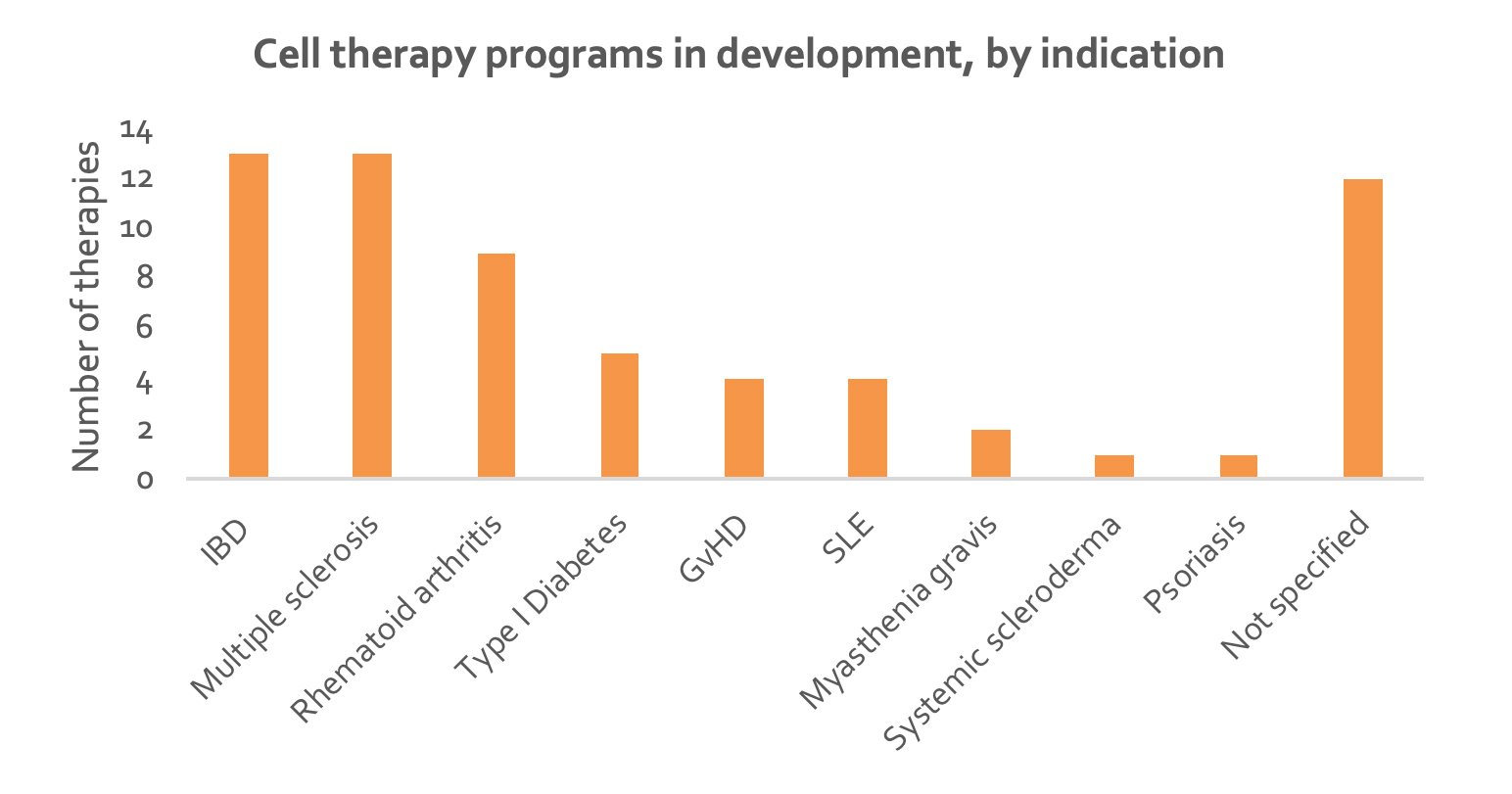
Figure 3. Column chart depicting number of cell therapies directed at each autoimmune disorder (data from individual companies). IBD, inflammatory bowel disease (includes ulcerative colitis and Crohn’s disease); GvHD, graft versus host disease; SLE, systemic lupus erythematosus.
MSCs
The potential of MSCs as a cell therapy has been investigated in several autoimmune disorders, including SLE, rheumatoid arthritis, multiple sclerosis and Crohn’s disease. Results have demonstrated safety as well as encouraging short-term clinical efficacy in all indications 21, 58, 59. However, most clinical trials have enrolled low patient numbers and long-term efficacy is yet unknown. In one study, a 5-year remission rate in SLE patients was reported at 34% 60.
Polyclonal Treg cells
Polyclonal Treg cells are being investigated in multiple sclerosis, Crohn’s disease, SLE and other undisclosed autoimmune disorders. Adoptive transfer of Tregs in clinical settings of GVHD and T1DM have proven to be safe and feasible and have demonstrated persistence for up to one year post-infusion 27, 61. However, the long-term efficacy of these therapies remains unproven.
CAR-T/Tregs
CAR-Tregs have been explored in murine models of colitis, multiple sclerosis and transplant rejection 62–64. They were shown to proliferate, secrete suppressive factors, and ameliorate disease symptoms in an antigen-specific manner. The first indications will likely be organ transplantation, where engineered Tregs targeting one antigen will yield short-term preliminary readouts for future complex autoimmune disorders. Indeed, Sangamo, which leads the field with TX200, is initiating clinical testing in kidney transplantation. TX200 is a Treg expressing a CAR against HLA-A2, gained through Sangamo’s acquisition of TxCell, to prevent rejection in HLA-2 mismatched kidney transplant patients, and is the first CAR-Treg candidate to be clinically tested 65.
Allogeneic versus autologous
70% of MSC therapies are being developed for off-the shelf administration (Figure 4). This is largely due to the difficulty and cost in isolating and expanding a large enough number of autologous MSCs. The popular use of allogeneic MSCs is based on their low immunogenicity, implying the possibility of greater availability via cell bank establishment 19. Indeed, several allogeneic MSC programs, such as from SCM Lifescience, offer a cell bank system. Celltex Therapeutics and Hope Biosciences also provide individual cell banks for their autologous candidates.
Autologous products are dominating in the Treg and CAR-T space. Persistence of Treg cells is of great importance, and will be challenging to achieve with an allogeneic product due to Treg cells’ complex editing pathway 66. Furthermore, the three-week time period of developing autologous treatments is not critical in autoimmunity 58. However, one needs to consider the risk of failure due to numbers of functionally defective Tregs. More importantly, the high cost associated with manufacturing and delivery is a challenge for health care systems. McGill, CEO of Quell Therapeutics, argues that the high price tag is justifiable if the engineered cells can persist long term 66.
It is likely that the science for allogeneic CAR-Tregs will follow from effector CAR-Ts in the oncology field. Sangamo is exploring the possibility of developing allogeneic Tregs using its zinc finger nuclease gene editing technology.
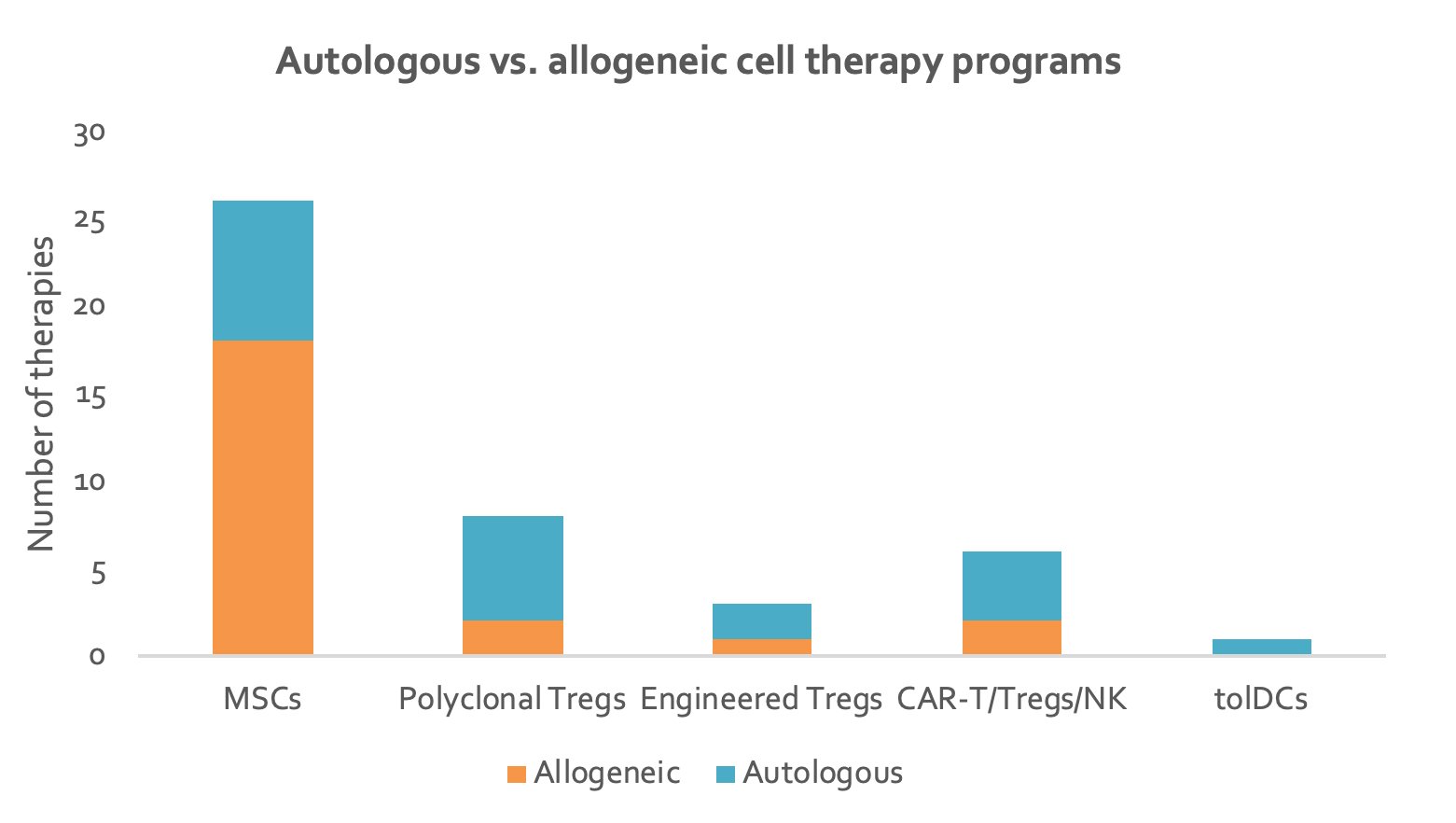
Figure 4. Stacked column chart depicting number of autologous and allogeneic cell therapies for each type of cell therapy (data from individual companies).
Efforts to enhance Treg cell specificity, survival and function
Although Treg cells are demonstrating promising clinical results, additional modifications will likely be needed to improve the cells’ potency and persistence. The field is exploring the use of a broad range of Treg-promoting therapies to use in combination with Treg therapy.
One strategy involves low-dose IL-2 to boost Treg number and function. Indeed, several groups are developing IL-2 mimics to promote Treg cell expansion in vivo. Sonoma has investigated the combination of low-dose IL-2 with polyclonal Tregs 67, suggesting that IL-2 can be incorporated into Treg therapy by developing engineered Tregs that secrete IL-2 and overexpress an IL-2 receptor on their surface 66.
An alternative strategy is to enforce expression of transcription factor FOXP3, which serves as a master regulator of Treg development and function, to promote persistence 26. Casebia Therapeutics has explored this strategy and is possibly developing FOXP3-overexpressing Treg cell therapies, but this has not been confirmed 66.
Optimising CAR-co-stimulatory endodomain design is being explored in the oncology field and has been exploited in next-generation CAR-T therapies. Whether expression of these co-stimulatory endodomains via CARs on Tregs enhances their function in a similar manner to those found on CAR-T cells is yet to be seen 58.
Safety
Overall, autoimmune disorders have a higher safety bar than cancer as they are not immediately life-threatening. Additionally, the risk of CAR-T-induced cytokine release syndrome in autoimmune disorders is significantly lower 66. However, the long-term immunosuppressive phenotype of Treg cells is unclear due to their high plasticity 34. Treg cells have shown to become unstable in murine models, whereby they lost expression of FOXP3 and converted to effector-like T cells 68. This problem is compounded by the chronic nature of autoimmune diseases, requiring Treg therapies to be long acting.
Several safety switch programs have been developed for CAR effector T cells to block potential adverse effects that could also be used in CAR-Tregs 26. For example, Bellicum Pharmaceuticals has developed CaspaCIDe, a safety switch technology. Alternatively, engineering CAR-Tregs to maintain FOXP3 expression could mitigate conversion 69.
Ultimately, the potential to alleviate these chronic diseases using cell-based therapies is enormous, with the field of autoimmune disease being one of the largest pharmaceutical markets in the world. Moreover, the quick evolution of CAR-T cells into the clinic in cancer has informed the community of the pitfalls associated with delivering an effective and safe treatment; applying these to CAR-Treg therapy will accelerate their clinical use. CAR-Tregs currently require further refinement including the need to maximize their suppressive function and stability, and better understand their homing capacity and persistency 58.
References:
1 M. D. Rosenblum, I. K. Gratz, J. S. Paw, and A. K. Abbas, “Treating human autoimmunity: current practice and future prospects.,” Sci. Transl. Med., vol. 4, no. 125, p. 125sr1, Mar. 2012, doi: 10.1126/scitranslmed.3003504.
2 “6. Autoimmune Diseases and the Promise of Stem Cell-Based | stemcells.nih.gov.” https://stemcells.nih.gov/info/2001report/chapter6.htm (accessed Jan. 04, 2021).
3 “Autoimmune Diseases: Current and Emerging Therapeutic Approaches.” https://www.labiotech.eu/reports/autoimmune-diseases-current-emerging-therapeutic-approaches/ (accessed Jan. 04, 2021).
4 C. J. Hawkey et al., “Autologous Hematopoetic Stem Cell Transplantation for Refractory Crohn Disease: A Randomized Clinical Trial.,” JAMA, vol. 314, no. 23, pp. 2524–2534, Dec. 2015, doi: 10.1001/jama.2015.16700.
5 L. Fugger, L. T. Jensen, and J. Rossjohn, “Challenges, Progress, and Prospects of Developing Therapies to Treat Autoimmune Diseases,” Cell, vol. 181, no. 1. Cell Press, pp. 63–80, Apr. 02, 2020, doi: 10.1016/j.cell.2020.03.007.
6 “Treatment Options for Autoimmune Disease.” https://www.news-medical.net/health/Treatment-Options-for-Autoimmune-Disease.aspx (accessed Jan. 04, 2021).
7 P. Li, Y. Zheng, and X. Chen, “Drugs for autoimmune inflammatory diseases: From small molecule compounds to anti-TNF biologics,” Frontiers in Pharmacology, vol. 8, no. JUL. Frontiers Media S.A., Jul. 12, 2017, doi: 10.3389/fphar.2017.00460.
8 G. Roda, B. Jharap, N. Neeraj, and J. F. Colombel, “Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management,” Clinical and Translational Gastroenterology, vol. 7, no. 1. Nature Publishing Group, p. e135, Jan. 07, 2016, doi: 10.1038/ctg.2015.63.
9 S. Chandrashekara, “The treatment strategies of autoimmune disease may need a different approach from conventional protocol: A review,” Indian Journal of Pharmacology, vol. 44, no. 6. Wolters Kluwer -- Medknow Publications, pp. 665–671, Dec. 2012, doi: 10.4103/0253-7613.103235.
10 J. S. Smolen, D. Aletaha, and I. B. McInnes, “Rheumatoid arthritis,” The Lancet, vol. 388, no. 10055. Lancet Publishing Group, pp. 2023–2038, Oct. 22, 2016, doi: 10.1016/S0140-6736(16)30173-8.
11 D. Wang et al., “Stem Cell Reports Ar ticle A Long-Term Follow-Up Study of Allogeneic Mesenchymal Stem/Stromal Cell Transplantation in Patients with Drug-Resistant Systemic Lupus Erythematosus,” Stem Cell Reports, vol. 10, pp. 933–941, 2018, doi: 10.1016/j.stemcr.2018.01.029.
12 T. Hlavaty et al., “Relapse rates of inflammatory bowel disease patients in deep and clinical remission after discontinuing anti-tumor necrosis factor alpha therapy.,” Bratisl. Lek. Listy, vol. 117, no. 4, pp. 205–211, 2016, doi: 10.4149/bll_2016_039.
13 M. Battaglia et al., “Rapamycin and interleukin-10 treatment induces T regulatory type 1 cells that mediate antigen-specific transplantation tolerance.,” Diabetes, vol. 55, no. 1, pp. 40–49, Jan. 2006.
14 M. Biswas et al., “Synergy between rapamycin and FLT3 ligand enhances plasmacytoid dendritic cell-dependent induction of CD41 CD251 FoxP31 Treg,” Blood, vol. 125, no. 19, pp. 2937–2947, 2015, doi: 10.1182/blood-2014-09-599266.
15 Q. Tang, “Therapeutic window of interleukin-2 for autoimmune diseases,” Diabetes, vol. 64, no. 6. American Diabetes Association Inc., pp. 1912–1913, Jun. 01, 2015, doi: 10.2337/db15-0188.
16 L. Ban, J. Zhang, L. Wang, W. Kuhtreiber, D. Burger, and D. L. Faustman, “Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism,” Proc. Natl. Acad. Sci. U. S. A., vol. 105, no. 36, pp. 13644–13649, Sep. 2008, doi: 10.1073/pnas.0803429105.
17 P. J. Eggenhuizen, B. H. Ng, and J. D. Ooi, “Treg Enhancing Therapies to Treat Autoimmune Diseases.,” Int. J. Mol. Sci., vol. 21, no. 19, Sep. 2020, doi: 10.3390/ijms21197015.
18 C. H. Mosanya and J. D. Isaacs, “Tolerising cellular therapies: what is their promise for autoimmune disease?,” Ann. Rheum. Dis., vol. 78, no. 3, pp. 297–310, Mar. 2019, doi: 10.1136/annrheumdis-2018-214024.
19 M. Lopez-Santalla, R. Fernandez-Perez, and M. I. Garin, “Mesenchymal Stem/Stromal Cells for Rheumatoid Arthritis Treatment: An Update on Clinical Applications,” Cells, vol. 9, no. 8. NLM (Medline), Aug. 07, 2020, doi: 10.3390/cells9081852.
20 R. J. Cheng et al., “Mesenchymal Stem Cells: Allogeneic MSC May Be Immunosuppressive but Autologous MSC Are Dysfunctional in Lupus Patients,” Frontiers in Cell and Developmental Biology, vol. 7. Frontiers Media S.A., p. 285, Nov. 15, 2019, doi: 10.3389/fcell.2019.00285.
21 T. Franceschetti and C. De Bari, “The potential role of adult stem cells in the management of the rheumatic diseases,” Therapeutic Advances in Musculoskeletal Disease, vol. 9, no. 7. SAGE Publications Ltd, pp. 165–179, Jul. 01, 2017, doi: 10.1177/1759720X17704639.
22 M. Liu et al., “Adipose-Derived Mesenchymal Stem Cells from the Elderly Exhibit Decreased Migration and Differentiation Abilities with Senescent Properties,” Cell Transplant., vol. 26, no. 9, pp. 1505–1519, Sep. 2017, doi: 10.1177/0963689717721221.
23 B. Lukomska, L. Stanaszek, E. Zuba-Surma, P. Legosz, S. Sarzynska, and K. Drela, “Challenges and Controversies in Human Mesenchymal Stem Cell Therapy,” Stem Cells International, vol. 2019. Hindawi Limited, 2019, doi: 10.1155/2019/9628536.
24 J. A. Ankrum, J. F. Ong, and J. M. Karp, “Mesenchymal stem cells: immune evasive, not immune privileged,” Nat. Biotechnol., vol. 32, no. 3, pp. 252–260, 2014, doi: 10.1038/nbt.2816.
25 A. J. Joswig et al., “Repeated intra-articular injection of allogeneic mesenchymal stem cells causes an adverse response compared to autologous cells in the equine model,” Stem Cell Res. Ther., vol. 8, no. 1, p. 42, Feb. 2017, doi: 10.1186/s13287-017-0503-8.
26 C. Raffin, L. T. Vo, and J. A. Bluestone, “Treg cell-based therapies: challenges and perspectives,” Nat. Rev. Immunol., vol. 20, no. 3, pp. 158–172, 2020, doi: 10.1038/s41577-019-0232-6.
27 J. A. Bluestone et al., “Type 1 diabetes immunotherapy using polyclonal regulatory T cells,” Sci. Transl. Med., vol. 7, no. 315, Nov. 2015, doi: 10.1126/scitranslmed.aad4134.
28 “CAR-T Cells Beyond Cancer: A New Therapy for Autoimmune Disease.” https://www.labiotech.eu/interviews/car-t-cells-txcell-treg-cells/ (accessed Jan. 04, 2021).
29 L. A. Stephens, K. H. Malpass, and S. M. Anderton, “Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg,” Eur. J. Immunol., vol. 39, no. 4, pp. 1108–1117, 2009, doi: 10.1002/eji.200839073.
30 Q. Tang et al., “In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes,” J. Exp. Med., vol. 199, no. 11, pp. 1455–1465, Jun. 2004, doi: 10.1084/jem.20040139.
31 P. Desreumaux et al., “Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease,” Gastroenterology, vol. 143, no. 5, 2012, doi: 10.1053/j.gastro.2012.07.116.
32 J. N. Clough, O. S. Omer, S. Tasker, G. M. Lord, and P. M. Irving, “Regulatory T-cell therapy in Crohn’s disease: Challenges and advances,” Gut, vol. 69, no. 5. BMJ Publishing Group, pp. 942–952, May 01, 2020, doi: 10.1136/gutjnl-2019-319850.
33 J. Rana and M. Biswas, “Regulatory T cell therapy: Current and future design perspectives,” Cellular Immunology, vol. 356. Academic Press Inc., p. 104193, Oct. 01, 2020, doi: 10.1016/j.cellimm.2020.104193.
34 E. W. Weber, M. V. Maus, and C. L. Mackall, “The Emerging Landscape of Immune Cell Therapies,” Cell, vol. 181, no. 1. Cell Press, pp. 46–62, Apr. 02, 2020, doi: 10.1016/j.cell.2020.03.001.
35 “Novel CAR NK-cell technology could lead to new treatments for lupus, other incurable diseases.” https://www.prnewswire.com/news-releases/novel-car-nk-cell-technology-could-lead-to-new-treatments-for-lupus-other-incurable-diseases-301057216.html (accessed Jan. 04, 2021).
36 M. P. Domogalla, P. V. Rostan, V. K. Raker, and K. Steinbrink, “Tolerance through education: How tolerogenic dendritic cells shape immunity,” Frontiers in Immunology, vol. 8, no. DEC. Frontiers Media S.A., p. 1764, Dec. 11, 2017, doi: 10.3389/fimmu.2017.01764.
37 S. Yamazaki, K. Inaba, K. V. Tarbell, and R. M. Steinman, “Dendritic cells expand antigen-specific Foxp3+CD25+CD4+ regulatory T cells including suppressors of alloreactivity,” Immunol. Rev., vol. 212, no. 1, pp. 314–329, Aug. 2006, doi: 10.1111/j.0105-2896.2006.00422.x.
38 “TiGenix and Takeda announce Alofisel receives approval in Europe.” https://www.takeda.com/newsroom/newsreleases/2018/tigenix-and-takeda-announce-alofisel-receives-approval-in-europe/ (accessed Jan. 04, 2021).
39 “NATIONAL INSTITUTE FOR HEALTH AND CARE EXCELLENCE Appraisal consultation document,” 2018.
40 “Mesenchymal Lineage Cells - Mesoblast Ltd.” https://www.mesoblast.com/science/mesenchymal-lineage-cells (accessed Jan. 04, 2021).
41 “Mesenchymal Stem Cells - hES-MSC / T-MSC-Welcome to Imstem.” http://www.imstem.com/abouten/show.php?lang=en&id=158 (accessed Jan. 04, 2021).
42 X. Wang et al., “Human ESC-derived MSCs outperform bone marrow MSCs in the treatment of an EAE model of multiple sclerosis,” Stem Cell Reports, vol. 3, no. 1, pp. 115–130, Jul. 2014, doi: 10.1016/j.stemcr.2014.04.020.
43 “Citius Pharmaceuticals Subsidiary NoveCite Announces Data from a Study of Induced Mesenchymal Stem Cell (‘i-MSC’) Therapy in an in vivo Model of Acute Respiratory Distress Syndrome (‘ARDS’).” https://www.prnewswire.com/news-releases/citius-pharmaceuticals-subsidiary-novecite-announces-data-from-a-study-of-induced-mesenchymal-stem-cell-i-msc-therapy-in-an-in-vivo-model-of-acute-respiratory-distress-syndrome-ards-301188345.html (accessed Jan. 04, 2021).
44 “SCM Lifescience.” http://www.scmlifescience.co.kr/en/business01.asp (accessed Jan. 04, 2021).
45 D. Mushahary, A. Spittler, C. Kasper, V. Weber, and V. Charwat, “Isolation, cultivation, and characterization of human mesenchymal stem cells,” Cytometry Part A, vol. 93, no. 1. Wiley-Liss Inc., pp. 19–31, Jan. 01, 2018, doi: 10.1002/cyto.a.23242.
46 “Our Technologies - Orbsen Therapeutics.” https://orbsentherapeutics.com/our-technologies/ (accessed Jan. 04, 2021).
47 “Opexa Therapeutics - Tcelna® - Tcelna® Description.” http://www.opexatherapeutics.com/tcelna/tcelna-description/default.html (accessed Jan. 04, 2021).
48 “GentiBio | We Make Tregs, Better.” https://www.gentibio.com/ (accessed Jan. 04, 2021).
49 “Descartes-08 CAR-T Cells in Generalized Myasthenia Gravis (MG) - Full Text View - ClinicalTrials.gov.” https://clinicaltrials.gov/ct2/show/NCT04146051 (accessed Jan. 04, 2021).
50 “Science | RNA-Armory - Cartesian.” https://www.cartesiantherapeutics.com/rna-armory/ (accessed Jan. 04, 2021).
51 “Regenerative Medicine | CRISPR.” http://www.crisprtx.com/programs/regenerative-medicine (accessed Jan. 04, 2021).
52 “Next Generation CAR T - Atara Biotherapeutics.” https://www.atarabio.com/science-and-technology/next-generation-car-t/ (accessed Jan. 04, 2021).
53 “China Immunotech (Beijing) Biotechnology Co. Ltd.-Technology & Product.” http://www.cn-immunotech.com/en/index/tech/cid/402.html (accessed Jan. 04, 2021).
54 Y. Chen, J. Sun, H. Liu, G. Yin, and Q. Xie, “Immunotherapy Deriving from CAR-T Cell Treatment in Autoimmune Diseases,” Journal of Immunology Research, vol. 2019. Hindawi Limited, 2019, doi: 10.1155/2019/5727516.
55 “Cabaletta raises $38M to develop CAR-T-like cell therapies for autoimmune diseases - MedCity News.” https://medcitynews.com/2018/11/cabaletta-raises-38m-to-develop-car-t-like-cell-therapies-for-autoimmune-diseases/?rf=1 (accessed Jan. 04, 2021).
56 “Technology - Cabaletta Bio.” https://cabalettabio.com/science/technology/ (accessed Jan. 04, 2021).
57 “Kyverna Therapeutics, Inc. | Taming Autoimmunity.” https://kyvernatx.com/ (accessed Jan. 04, 2021).
58 Y. R. Mohseni, S. L. Tung, C. Dudreuilh, R. I. Lechler, G. O. Fruhwirth, and G. Lombardi, “The Future of Regulatory T Cell Therapy: Promises and Challenges of Implementing CAR Technology,” Frontiers in Immunology, vol. 11. Frontiers Media S.A., p. 1608, Jul. 24, 2020, doi: 10.3389/fimmu.2020.01608.
59 M. Carvello, A. Lightner, T. Yamamoto, P. G. Kotze, and A. Spinelli, “Mesenchymal Stem Cells for Perianal Crohn’s Disease,” Cells, vol. 8, no. 7, p. 764, Jul. 2019, doi: 10.3390/cells8070764.
60 D. Wang et al., “A Long-Term Follow-Up Study of Allogeneic Mesenchymal Stem/Stromal Cell Transplantation in Patients with Drug-Resistant Systemic Lupus Erythematosus,” Stem Cell Reports, vol. 10, no. 3, pp. 933–941, Mar. 2018, doi: 10.1016/j.stemcr.2018.01.029.
61 C. G. Brunstein et al., “Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: Safety profile and detection kinetics,” Blood, vol. 117, no. 3, pp. 1061–1070, Jan. 2011, doi: 10.1182/blood-2010-07-293795.
62 E. Elinav, N. Adam, T. Waks, and Z. Eshhar, “Amelioration of Colitis by Genetically Engineered Murine Regulatory T Cells Redirected by Antigen-Specific Chimeric Receptor,” Gastroenterology, vol. 136, no. 5, pp. 1721–1731, 2009, doi: 10.1053/j.gastro.2009.01.049.
63 M. Fransson et al., “CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery,” J. Neuroinflammation, vol. 9, no. 1, p. 576, May 2012, doi: 10.1186/1742-2094-9-112.
64 K. G. MacDonald et al., “Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor,” J. Clin. Invest., vol. 126, no. 4, pp. 1413–1424, Apr. 2016, doi: 10.1172/JCI82771.
65 “Sangamo Launching First-in-human Clinical Trial Testing TX200 Therapy.” https://alportsyndromenews.com/2019/11/25/sangamo-to-launch-first-in-human-clinical-trial-testing-tx200-cell-therapy-in-end-stage-renal-disease/?cn-reloaded=1 (accessed Jan. 04, 2021).
66 “T regs are back – promising to do for autoimmunity what CAR Ts have done in cancer – QuellTX.” https://quell-tx.com/t-regs-are-back-promising-to-do-for-autoimmunity-what-car-ts-have-done-in-cancer/ (accessed Jan. 04, 2021).
67 “T1DM Immunotherapy Using Polyclonal Tregs + IL-2 (TILT),” [Online]. Available: https://clinicaltrials.gov/ct2/show/NCT02772679.
68 X. Zhou et al., “Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo,” Nat. Immunol., vol. 10, no. 9, pp. 1000–1007, 2009, doi: 10.1038/ni.1774.
69 C. R. Maldini, G. I. Ellis, and J. L. Riley, “CAR T cells for infection, autoimmunity and allotransplantation,” Nature Reviews Immunology, vol. 18, no. 10. Nature Publishing Group, pp. 605–616, Oct. 01, 2018, doi: 10.1038/s41577-018-0042-2.