Traditional drug development focuses on targeting proteins with a defined binding pocket or active site to regulate protein activity. These strategies have exploited only around 400 of the 4000 disease-associated proteins 1, which means 90% of disease-associated proteins, including many high priority targets, currently remain ‘undruggable’ 2. However, a new pharmacology paradigm is on the horizon: targeted protein degradation (TPD).
Leveraging the ubiquitin-proteasome system
TPD typically utilizes the ubiquitin-proteasome system (UPS) to selectively regulate biological pathways by degrading a target protein through its association with a particular ubiquitin ligase (E3). The human genome encodes over 600 E3 ligases and each targets a specific subset of proteins. The most widely used E3 ligases include Von Hippel-Lindau (VHL), cereblon (CRBN), cellular inhibitor of apoptosis proteins (cIAPs), and mouse double minute 2 homolog (MDM2), which have all been used to degrade proteins via proteolysis-targeting chimeras (PROTAC) or via molecular glues. MDM2 and CRBN have demonstrated this in the clinic, VHL in vivo and cIAPs in vitro. VHL and cIAPs are currently being investigated in the clinic. Their characterization has highlighted potent binding motifs for other molecules such as bestatin derivatives for cIAPs, nutlin derivatives for MDM2, peptidomimetics of HIF1a binding peptide for VHL, and certain clinical anti-cancer immunomodulatory drugs (such as thalidomide or lenalidomide) for CRBN 3. Indeed, lenalidomide’s (sold as Revlimid by Celgene) ability to recruit UPS through CRBN for selective degradation of Ikaros family zinc finger proteins 1 and 3 (IKZF1 and 3) has been shown to be necessary for its therapeutic effect in multiple myeloma both in vivo and in vitro 4. Lenalidomide’s mechanism was only elucidated in 2014; 14 years after its launch in 2000.
An understanding of the structural biology of E3 ligases has subsequently contributed to the development of the heterobifunctional activity of PROTACs and molecular glues. Additionally, it has helped provide transformative treatments to patients with multiple myeloma and erythema nodosum leprosum for example 5. It has also helped pave the way for TPD by highlighting the clinical potential of drugs that manipulate protein homeostasis while also demonstrating that TPD drug strategy is both clinically and commercially viable.
PROTACs and molecular glues are the most advanced TPD technologies and have the potential to minimise drug resistance, reduce toxicity and regulate functions not dependent on enzyme action through their increased selectivity and potency. Moreover, they provide access to intracellular proteins which small molecule inhibitors have limited access to 1.
Types of TPD technologies
The first proof-of-concept (PoC) TPD approach was seen in 2001 in the form of a peptide-based PROTAC that targeted the enzyme methionine aminopeptidase-2 (MetAP-2; thought to play an important function in human endothelial cell proliferation, and as such provides a valuable target in both inflammation and cancer). Seven years later, a small molecule PROTAC that could bind the E3 ligase MDM2 to degrade androgen receptors was developed 6. Then followed molecular glues, after which the TPD space started expanding into usage of lysosomes and autophagy, increasing the range of proteins that could be targeted. This was largely because the UPS system could only target soluble and short-lived proteins while the lysosomal pathway could target insoluble, long-lived and membrane proteins 1. For example, GlueTAC is a covalent nanobody (GlueBody) conjugated with a cell-penetrating peptide and a lysosomal-sorting sequence, resulting in a GlueTAC chimera. It allows cross-linking with surface antigens on cancer cells based on proximity and therefore membrane protein degradation 7. Researchers at Peking University, China showed that this nanobody-based PROTAC strategy has higher efficacy than atezolizumab (Tecentriq); the latter selectively binds to PD-L1 to stop the interaction between PD-1 and B7.1 (i.e., CD80 receptors) and is a first-line treatment for PD-L1 positive tumors 8. The study showed a higher degradation ratio of PD-L1, and significant T-cell activation enhancement compared to Tecentriq in a non-small cell lung cancer (NSCLC) cell line. Moreover, GlueTAC inhibited tumor growth in the immunodeficient NOD/Shi-scid/IL-2Rγ (NOG) mouse (a new generation of a severely immunodeficient mouse) whereas atezolizumab only slowed tumor growth in the same animal model 7. However, lysosome and similar-based technologies are still in their infancy as their mechanisms of action have yet to be fully elucidated.
PROTACs are heterobifunctional molecules which use a linker to connect an E3 ligase with a target protein to form a ternary complex, thereby interacting with both proteins simultaneously. Similarly, molecular glues also form this ternary complex, but only interact with a single protein (E3 or target) instead of both, and act by stabilizing the protein-protein interactions between the ligase and target. This connection induces ubiquitylation of the target leading to its degradation by the proteasome 9. These drugs are then recycled by disassociating from the ternary complex after the target has been degraded. Consequently, they are free to form a new complex with another E3 ligase and a new target to induce another catalytic cycle. This catalytic mechanism means that they act in a sub-stoichiometric manner, which reduces dosing and potential side effects 2.
Challenges in drug development
Both drug groups have advantages and disadvantages. While PROTACs struggle with cell permeability, the smaller molecular glues generally have greater oral bioavailability, but are more difficult to rationally design. With both, the degradation of whole proteins instead of inhibition can lead to both greater on-target and off-target toxicities and it is still unclear how exploiting UPS affects the organism as a whole 1. Further complicating matters is the fact that there is no specific guidance on how the traditional drug metabolism and pharmacokinetics model (DMPK) can be optimised for TPD drug development, and conventional PK screening and in vitro permeability evaluation systems are not suitable for PROTACs. Their unique structures also mean that they generally will not meet the typical parameters for drug-likeness and oral availability evaluation tools such as Lipinski’s Rules. Additionally, PROTAC drugs are intrinsically prone to the hook effect at concentrations of 1-10uM (which is the range used to assess small molecule drug efficacy) 10 i.e., at high concentrations, a target protein-PROTAC or E3 ligase-PROTAC binary complex is formed competitively, leading to reduced efficacy or toxic reactions 11. In addition to these pharmacokinetic challenges, the endogenous concentration of the E3 ligase also plays a role in drug efficacy. For example, MS-140, a preclinical stage PROTAC under development by Eli Lilly & Co degrades cyclin-dependent kinase 4 and 6 (CDK4 and 6) in vitro with potency depending on endogenous expression of CRBN. Although this may limit which tissues and cancers respond to treatment, it can also improve diagnosis and allow for localized therapy 12.
These unique challenges can somewhat be mitigated through early screening of in vitro characteristics, optimizing solubility and metabolite clearance, and using the molecules with the best oral bioavailability for downstream pharmacokinetic studies 11 (some scientists have developed more useful albeit early-stage approaches to determining oral availability of PROTACs via LC-MS/MS 13. Molecular glues such as rapamycin, thalidomide and pomalidomide are already on the market, demonstrating that progressing this drug type through development and all the way to the patient is indeed achievable. Interestingly, the latter mentioned drugs were approved by the FDA before their mechanisms were elucidated 1. Whereas thalidomide and pomalidomide act by interacting with CRBN to induce degradation of the target through UPS, rapamycin induces degradation of mTOR by bypassing ubiquitination. In both an in vivo and in vitro study, rapamycin’s stabilisation of the FKBP12-rapamycin-FRB (mTOR) ternary complex induced its localization to the proteasome and consequently its degradation 14. This aligns with its role as a regulator of proteasomal function and allosteric inhibiting function of mTOR 15. Interestingly, although rapamycin is described as a molecular glue by some, others would argue that as it does not induce degradation via E3 ligases nor the UPS system, it does not strictly fall into this drug category.
Other TPD technologies
Another form of TPD technology is the lysosome-based TPD autophagy-targeting chimera (AUTOTAC), which has been shown to remove protein aggregates such as misfolded tau. It specifically targets the P301L tau mutant, which has prion-like behavior, resulting in tau degradation and thereby removing neurofibrillary tangles 16 involved in Alzheimer’s disease. Another lysosome-based TPD, ATTEC can selectively degrade mutant Huntington proteins (mHTT) through its interaction with expanded polyQ stretches (absent in the wild-type) and the autophagosome protein microtubule-associated protein 1A/1B light chain 3 (LC3). This ATTEC can also promote degradation of other disease-associated proteins that contain the elongated polyQ sequence 17. For example, ATTECs can also target BTK, which plays a prominent role in cancer. PROTACs have also demonstrated efficacy in this area, as is evident by Nutrix Therapeutics’ BKT-targeting PROTAC degrader, NX-2127. Typically, mutations render conventional inhibitors ineffective, however TPD drugs are still able to target mutant BTK proteins 1.
The TPD drug pipeline is in its infancy
The TPD drug landscape is young but growing. There are approximately 110 TPD drugs in development, with around 51 in the clinic (see Figure 1) 5,18,19,20.
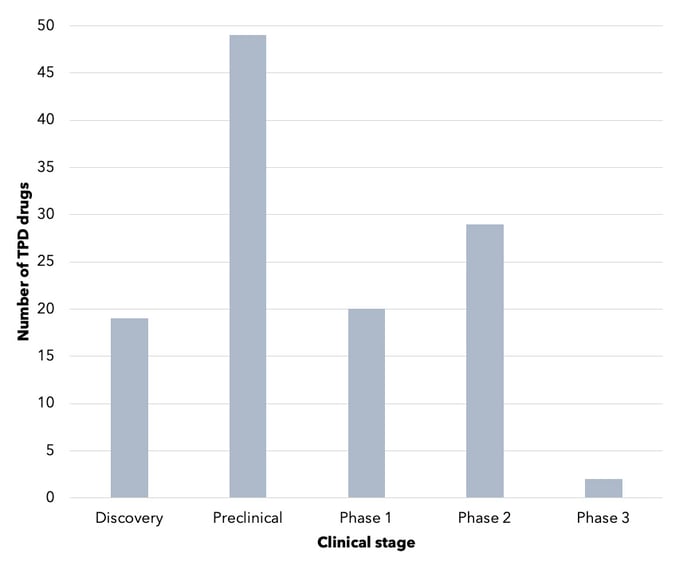
Figure 1. Number of TPD drugs, by stage of development. This is not an exhaustive representation of the TPD industry. Source: Cortellis.
Of these, 25 are PROTAC drugs being developed mostly for cancer, but also to treat neurological disorders (see Figure 2) 20. Some of these drugs target multiple indications, such as the University of Michigan’s ARD-61 which targets both androgen receptor-positive breast and prostate cancers 21. Suzhou Kintor Pharmaceuticals’ GT-20029 is being developed for both androgenetic alopecia (AGA) and acne (via a topical treatment for both). Both are conditions that have a large unmet need with limited treatments available 22.
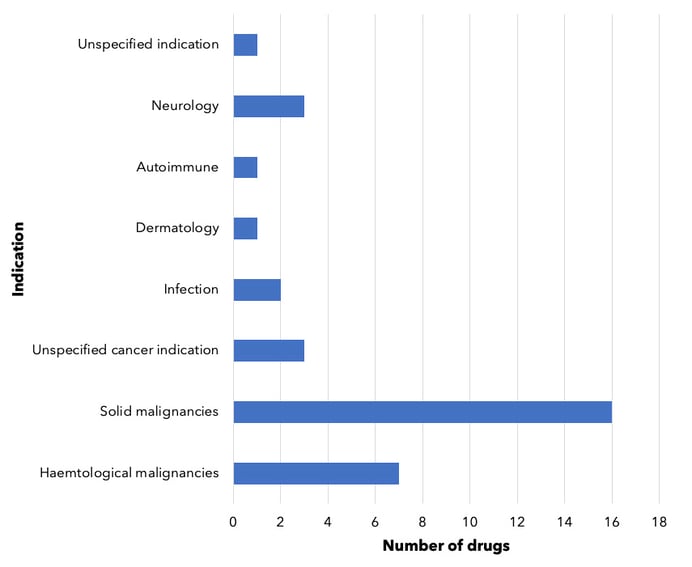
Figure 2. PROTAC drugs in development, by indication. Drugs being developed for multiple indications are counted multiple times, as appropriate. For drugs counted under 'unspecified cancer indication’ and ‘unspecified indication’, companies have not yet disclosed specific indications for these drugs. Dermatologic branch includes androgenetic alopecia as well as acne.
In the neurological disorders space, PROTAC drugs may offer advantages over conventional treatments. In addition to their superior catalytic activity, they can also cross the blood-brain barrier and enter cells of the central nervous system (CNS). This was demonstrated with Arvinas’ PROTAC protein degrader, a tau targeting PROTAC candidate which reduced pathological tau by over 95% in the murine pathologic tau model (P301L) 23.
The most popular target for PROTACs seems to be the androgen receptor (with three drugs in development), followed by Bruton’s tyrosine kinase (BTK), hematopoietic progenitor kinase 1 (HPK1), and cancerous K-RAS mutations (refer to Figure 3).
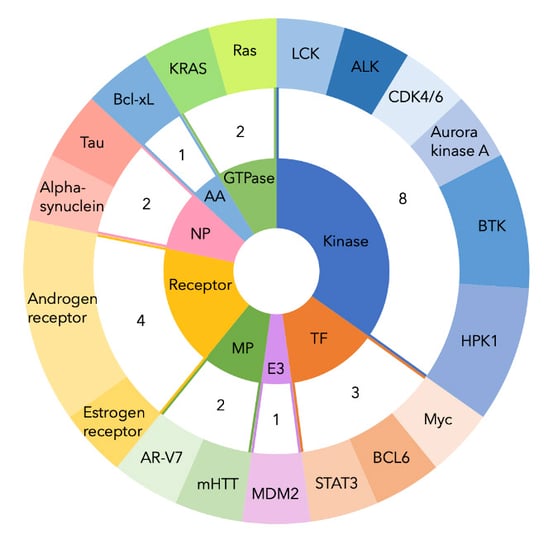
Figure 3. PROTAC drugs in development, by target. Two-tiered doughnut chart showing number of PROTAC drugs in development, grouped by target and color-coded by protein class. NP, neuronal protein. AA, anti-apoptotic protein. TF, transcription factor. E3, E3 ligase. MP, mutant protein. ALK, anaplastic lymphoma kinase. LCK, lymphocyte-specific protein tyrosine kinase. BCL6, B cell lymphoma 6. STAT3, signal transducer and activator of transcription 3. AR-V7, androgen receptor splice variant 7. mHTT, mutant huntingtin protein. Bcl-xL, B-cell lymphoma extra-large.
Despite these challenges, the TPD opportunity is gaining interest from investors and drug developers. Between $3.5 - 5 billion has been estimated to have been invested in the TPD area since 2014. Deal volume doubled from seven deals in 2018 to 16 in 2020-2021 24. This heightened interest in the space is thought to have arisen after Arvinas, a leading PROTAC company, presented promising efficacy and safety data for two drugs, ARV-471 and ARV-110. ARV-471 is an oestrogen receptor (ER) degrader for the treatment of breast cancer, while ARV-110 targets metastatic castration-resistant prostate cancer 25. In December 2021, Arvinas presented efficacy and safety data for ARV-471 from a phase II clinical trial in patients with locally advanced or metastatic ER+/HER2- breast cancer that had received prior chemotherapy. The results showed ER degradation of up to 89% in these patients, with a median degradation of 67%. The clinical benefit rate was 40% 26. This is comparable to the clinical benefit rate of 38.7% of tamoxifen 27, which is currently the first-line treatment for all stages of ER positive breast cancer 28. ARV-110 also yielded impressive results with a reduction of prostate-specific antigen levels of ³50% in 46% of patients with the AR T878X and/or H875Y mutations 29.
Arvinas is the leading specialist biotech firm with other specialist firms including Vividion Therapeutics, C4 Therapeutics, Nutrix Therapeutics, Kymera and Evotec. Similarly to Arvinas which is a spin out from Yale University, Vividion Therapeutics is a spin from Scripps Research Institute. Vividion uses its own proprietary chemoproteomic platform technology to overcome the limitations of traditional screening methods to scrutinize new functional characteristics of proteins that can be exploited for downstream drug development. Having established partnerships with Roche and Celgene amounting to a sum of $236m, and its recent acquisition by Bayer for an upfront payment of $1.5bn and $500m in milestone payments, Vividion seems poised for success 30,31. C4 Therapeutics uses its own proprietary TORPEDO (target-oriented protein degrader optimizer) platform to develop molecular glues and has collaborations with Roche and Biogen. Nutrix Therapeutics also deploys its own DELigase platform to search for molecules that can both induce and prevent protein degradation. Kymera takes a slightly different approach by using their Pegasus platform together with computational characterization, to identify the expression profile of ~600 E3 ligases across different tissues to pair up ligases with appropriate target proteins 5.
Bristol Myers Squibb (BMS) currently dominates the market with an impressive pipeline that covers a large range of indications and TPD drugs. This was made possible through its acquisition of Celgene, which originally marketed the immunomodulatory CRBN-binding drugs. For example, the annual sales of Celgene’s Revlimid (lenalidomide) grew from $5bn in 2014 to $12bn in 2020, making it the third most sold drug in the world in 2020 5. Additionally, BMS entered a collaboration with Evotec, which will last into 2030 based on developing molecular glue degraders by leveraging Evotec’s PanOmics platform (32). This agreement includes an upfront payment of $200m with additional milestone payments, and a potential of $5bn royalties on sales 33. These specialist firms are all leveraging their own proprietary protein screening/development platform to develop putative degraders in the TPD space. Evidently, this suggests that to excel within this space that aims to address the undruggable, novel technology and screening methods are an essential element of the recipe to success. Major pharma companies such as BMS are forming partnerships with these specialist firms to gain greater potential market share. Other companies following BMS’ footsteps include Pfizer, Sanofi and Novartis 5. Despite a low barrier to entry in the TPD industry now that the putative binding motifs of key E3 ligases have been elucidated, the medicinal chemistry required to create a functional drug with superior efficacy to conventional treatment remains challenging 5.
The TPD market has seen many partnerships formed as smaller biotech companies seek to leverage the commercial and clinical expertise of bigger pharma companies. In May 2022, Bristol Myers Squibb entered a collaboration with Amphista to leverage its EclipsysTM platform, with up to $1.5bn in milestone payments 34 – the platform is used for the discovery and development of target protein therapeutics. Another company, Kymera, has recently raised $150m (August 2022) to support its target protein degradation platform ‘Pegasus’ – the platform exploits the UPS system and helps cells target potential disease-associated proteins 35.
Conclusion
The commercial opportunity for biotech and pharmaceutical companies in this space is large given the vast number of disease-associated proteins that TPD technology can theoretically target. Approximately 400 of the 600 proteins thought to play a role in cancer are not enzymes and therefore do not possess an active site nor a sufficient hydrophobic pocket for conventional small molecule inhibitors to target - this opens the door for PROTAC drugs 5. There are currently over 25 specialized technology platforms to facilitate PROTAC development and many established fragment-based discovery and covalent fragment discovery platforms. As these continue to become more sophisticated, TPD has the potential to disrupt the industry of conventional therapies and usher in a new age of drug therapy. Proteins that were previously considered undruggable could become druggable, and drug resistance mechanisms that currently limit clinical utility could become less common 2.
References
2 Martín-Acosta, P., & Xiao, X. (2021). PROTACs to address the challenges facing small molecule inhibitors. European Journal of Medicinal Chemistry, 210.
3 Chopra, R., Sadok, A. & Colns, I., 2019. A critical evaluation of the approaches to targeted protein degradation for drug discovery. Drug Discovery Today: Technologies, Volume 31, pp. 5-13.
4 Lu, G. et al., 2013. The Myeloma Drug Lenadomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science, 343(6168), pp. 305-309.
5 Shimmings, A. (2022). Scrip’s Rough Guide To Targeted Protein Degradation. Informa Pharma Intelgence.
6 Alabi, S. B., & Crews, C. M. (2021). Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. Journal of Biological Chemistry, 296.
7 Zhang, H. et al., 2021. Covalently Engineered Nanobody Chimeras for Targeted Membrane Protein Degradation. Journal of the American Chemical Society, 143(40), p. 16377–16382.
8 Herbst, R. et al., 2020. Atezozumab for First-ne Treatment of PD-L1–Selected Patients with NSCLC. The New England Journal of Medicine, Volume 383, pp. 1328-1339.
9 Békés, M., Langley, D. R., & Crews, C. M. (2022). PROTAC targeted protein degraders: the past is prologue. Nature Reviews Drug Discovery, 21, 181–200.
10 Cecchini, C., Pannilunghi, S., Tardy, S., & Scapozza, L. (2021). From Conception to Development: Investigating PROTACs Features for Improved Cell Permeabity and Successful Protein Degradation. Frontiers in Chemistry.
11 Ma, L. (2022, April 15). Tackng the DMPK challenges of developing PROTAC drugs. Drug Discovery & Development.
12 Caksa, S. & Apn, A. E., 2021. PROactively TACkng CDK4/6 therapy resistance. Nature Cancer, Volume 2, p. 372–373.
13 Nguyen, T.-T.-L., Kim, J. W., Choi, H.-I., Maeng, H.-J., & Koo, T.-S. (2022). Development of an LC-MS/MS Method for ARV-110, a PROTAC Molecule, and Appcations to Pharmacokinetic Studies. Molecules, 27(6).
14 Janse, D. M., Crosas, B., Finley, D. & Church, G. M., 2004. Locazation to the Proteasome Is Sufficient for Degradation. Journal of Biological Chemistry, 279(20), pp. 21415-21420.
15 Osmulski, P. A. & Gaczynska, M., 2013. Rapamycin allosterically inhibits the proteasome. Molecular Pharmacology, 84(1), pp. 104-13.
16 Ji, C., Kim, H., Lee, M. & al., e., 2022. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nature Communications, Volume 13.
17 , Z. et al., 2020. ATTEC: a potential new approach to target proteinopathies. Autophagy, Volume 16, p. 185–187.
18 Report nker. (2021). Targeted Protein Degradation Market: Focus on Technology Platforms and Therapeutics, 2021-2030: Distribution by Type of Protein degrader, Therapeutic Area, Route of administration, Key Contributing Technologies and Key Geographical Regions. Retrieved August 9, 2022, from https://www.globenewswire.com/news-release/2021/07/08/2259727/0/en/Targeted-Protein-Degradation-Market-Focus-on-Technology-Platforms-and-Therapeutics-2021-2030-Distribution-by-Type-of-Protein-degrader-Therapeutic-Area-Route-of-administration-Key-C.html
19 Drug Discovery World. (2022, January 24). Why the emergence of targeted protein degradation is so exciting. Drug Discovery World.
20 Cortels. (2022). Retrieved August 19, 2022, from https://www.cortels.com/intelgence/qsearch/protac?indexBased=true&searchCategory=CI
21 Zhao, L., Han, X., Lu, J., McEachern, D., & Wang, S. (2020). A highly potent PROTAC androgen receptor (AR) degrader ARD-61 effectively inhibits AR-positive breast cancer cell growth in vitro and tumor growth in vivo. Neoplasia, 22(10), 522–532.
22 PR Newswire. (2022, August 9). Kintor Pharma Announces Completion of Subject Enrollment and Dosing in Phase I Cnical Trial of AR-PROTAC(GT20029) in China. PR Newswire.
23 Globe Newswire. (2019, July 18). Arvinas to Present Precnical Tau-Directed PROTAC® Protein Degrader Data at Alzheimer's Association International Conference. Arvinas Press Release.
24 Nasir, M., Bak, P., Gauldie, S., & Leo, C. (2022). From drug target inhibition to degradation: a TACtical strategy. Biopharma Dealmakers.
25 Arvinas, 2019. Arvinas Presents a Platform Update, Including Initial Data from the First Two Cnical Trials of PROTAC® Targeted Protein Degraders. Arvinas Press Release, 23 October.
26 (2021, December 10). Arvinas and Pfizer Announce PROTAC® Protein Degrader ARV-471 Continues to Demonstrate Encouraging Cnical Benefit Rate in Patients with Locally Advanced or Metastatic ER+/HER2- Breast Cancer. Pfizer Press Release.
27 Arpino, G., Krishnan, M. N., Dinesh, C. D., Bardou, V., Clark, G., & Elledge, R. (2003). Idoxifene versus tamoxifen: a randomized comparison in postmenopausal patients with metastatic breast cancer. Original Article Breast Cancer, 14(2), 233-241.
28 McDonnell, D. P., Wardell, S. E., & Norris, J. D. (2015). Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. Journal of Medicinal Chemistry, 58(12), 4883–4887.
29 Globe Newswire. (2022, February 22). Arvinas PROTAC® Protein Degrader Bavdegalutamide (ARV-110) Continues to Demonstrate Cnical Benefit in Men with Metastatic Castration-Resistant Prostate Cancer. Arvinas Press Release.
30 Al Idrus, A., 2020. Roche taps Vividion in $135M protein degradation deal. Fierce Biotech, 19 May.
31 Vividion Therapeutics, 2021. Bayer Strengthens Drug Discovery Platform Through Acquisition of Vividion Therapeutics. Vividion Therapeutics Press Release, 5 August.
32 Evotec, 2021. Bristol Myers Squibb exercises option to extend targeted protein degradation partnership with Evotec. Evotec Press Release, 26 April.
33 Evotec, 2022. Evotec and Bristol Myers Squibb extend and expand strategic partnership in protein degradation. Evotec Press Release, 10 May.
34 Amphista Therapeutics. (2022, May 4). Amphista Therapeutics Enters Strategic Collaboration with Bristol Myers Squibb for Discovery and Development of Targeted Protein Degradation Therapeutics. Amphista Therapeutics.
35 Keown, A. (2022, August 19). Kymera Raises $150 Milon to Support Protein Degradation Program. BioSpace.