Intro:
Animal modelling is a staple of preclinical drug development and in most countries a legal requirement before entering human trials. However, these experiments are time-consuming, expensive, and rarely provide adequate translational power, particular in disease modelling and efficacy studies. Unnecessary use of animal testing further extends not only the cost and timelines of reaching the clinic, but also wastes animal life and endangers trial subjects. We take this opportunity to review common pitfalls in animal testing and subsequent translational failures. We discuss the ongoing considerations for animal testing, particularly for early-stage research, and highlight some of the emerging technologies that could optimise translational research in future. In particular, we cover methods and techniques to optimise preclinical testing within the context of the 3Rs categorisation.
First proposed by Russell and Burch in 1959, the 3Rs provide a strategy for Reduction, Refinement, and Replacement in animal testing, and are an internationally accepted set of principles incorporated into legislations, guidelines and practice of animal experiments to safeguard animal welfare [1], [2].
- Reduction - methods that minimise the number of animals used per experiment, or methods which allow the information gathered per animal in an experiment to be maximised, to reduce the use of additional animals.
- Refinement - methods that minimise the pain, suffering, distress, or lasting harm that may be experienced by research animals in aspects of animal use, from their housing and husbandry to the scientific procedures performed on them.
- Replacement - technologies or approaches which directly replace or avoid the use of animals in experiments where they would otherwise have been used.
Use of animal models in safety and pharmacology testing remains vital, and although there are alternative emerging technologies which can provide early insight into safety and toxicity, animal models will likely remain the gold standard for the foreseeable future. However, history has delivered multiple examples of failure to predict human toxicity and the quantifiable shift in reducing the use of animals in toxicology testing has been shown [3].
Our primary discussion focuses on the applications of the 3Rs in pathophysiology and efficacy animal modelling in drug development. Contrary to safety assessment, the evaluation of efficacy is not subject to formalised guidance or regulations as each new drug warrants a tailor-made approach based on its mechanism of action and indication.
The predictive power of animal models
Predictive validity is a paramount goal in modelling disease; however, this can only be evaluated retrospectively. Given the continually high attrition rate for therapeutic products, we could confidently state that animal models are not currently fully predictive of human response [4].
Broadly speaking, animal models are used in two ways: firstly, to generate (exploratory) and test (confirmatory) hypotheses and secondly, to predict outcomes in humans. The first is applied earlier in preclinical development to study pathophysiology and formulate proposals on how best to therapeutically target disease. The second is used in later stages where a mode of activity has been established and animal models are used to select leads and generate data intended (hoped) to be predictive of human response. Understandably, despite primates representing the closest human experience, mice and rats are the most common species used given the lower ethical hurdle and expense.
There continue to be significant limitations in representing human disease and a lack of validated preclinical models in multiple therapeutic areas. An AstraZeneca study assessing its failure rates for small molecule programs showed that 40% of clinical efficacy failures were linked back to preclinical modelling issues, including lack of validated disease models [5]. Research within the field of psychiatric disorders led to the classification and definition of criteria for animal model validation; these principles can be applied to most areas of therapeutic development [6], [7]. The first component is face validity (similarity to the modelled condition); the second is construct validity (the model has a sound theoretical rationale); and the third, predictive validity (prediction of efficacy in the clinic).
By therapeutic area
Wong et al recently published a large-scale study summarising clinical success rates across different therapeutic areas [8]. As expected, oncology fared the worst with an overall success rate of 3.4% from Phase I to approval, followed by CNS (15.0%) and autoimmune/inflammation (15.5%). Given the particularly poor success rates in oncology, CNS, autoimmune and inflammation, we focus our discussion on the contributions of preclinical modelling in these areas.
Oncology
Tumours and their environments are made up of heterogeneous malignant cells, normal and abnormal stroma, immune cells, and microenvironment containing chemokines, cytokines, and growth factors. The genetic, epigenetic, and environmental drivers of cancer development complicate preclinical modelling. Cancer is dynamic, it grows, mutates, metastasises, and often evolves mechanisms of immune system and drug resistance. Atypical disease aspects like pseudoprogression, mostly seen in immunotherapy trials as the appearance of new lesions or increase in primary tumor size followed by tumor regression, has not been well-described in cell or murine models [9]. Moreover, clinical endpoints in cancer are defined mainly in terms of patient survival rather than the intermediate endpoints seen in other disciplines. Therefore, it takes many years before the clinical applicability of initial preclinical observations can be known.
In a recent lung cancer study, the preclinical data of 134 failed lung cancer drugs and 27 successful lung cancer drugs were correlated with approval outcome [10]. The authors found that comparing success of drugs according to tumour growth inhibition produced interval estimates too wide to be statistically meaningful and therefore concluded no significant linear trend between preclinical success and drug approval. For example, from 378 in vitro cell culture experiments, the mean logIC50 (nM) value for approved drugs was 2.94 and for failed was 3.70 with a p value of only 0.216. Likewise, from 144 preclinical mouse experiments the median tumour growth inhibition (TGI) % for approved drugs was 75% and failed was 77% with p=0.375.
Subcutaneous and xenograft models are the simplest and most used animal models in anticancer drug development. However, these models generally have poor resemblance to a natural in vivo tumour. Subcutaneous models for example lack an established microenvironment, are often highly vascularised allowing for fast clearance, and limit the development of metastases. Orthotopic models improve the representation of microenvironment. However, if over manipulated in vitro, inoculated cells in xenograft models may lose tumour histology and heterogeneity [7]. Moreover, most models (apart from patient-derived xenografts) are based on clonal/semi-clonal tumour cell lines that do not represent the heterogeneity of tumours. Guerin et al conducted a detailed comparison of transplanted and spontaneous tumour animal models, focussing on structure–function relationships in the tumour microenvironment [11]. The analysis highlighted multiple key points of differentiation between the two. For example, the growth of transplanted tumours slows down with time, yet growth of spontaneous tumours is initially slow and then accelerates with time. They demonstrate a differential drug effect showing that anti-cancer drug 5,6-dimethylxanthenone-4-acetic acid induced a transient regression of transplanted melanomas but not of isogenic spontaneous tumours.
The consequences of failing to understand these limitations in animal models can be significant. Take, for example, the evaluation of sunitinib (Pfizer) in treating breast cancer [12]. Sunitinib showed promising preclinical results in multiple breast cancer models, including typical xenograft, transgenic, and chemically induced. Yet it failed to meet survival endpoints in four separate Phase III trials of metastatic breast cancer (including chemotherapy combination). A subsequent study by Guerin et al showed that the reason for this failure was owed to the poor representation of metastatic disease in the preclinical models selected. The authors showed that in a more reliable model of post-surgical metastatic breast cancer, there was indeed no survival benefit with sunitinib, the only impact was on orthotopic primary tumours.
CNS
CNS is a notoriously complex disease area and perhaps the most challenging to represent in animal models. The diverse nature of disease establishment ranges from genetic (ataxias and lysosomal storage disorders) through to protein misfolding and aggregation (prions), and those of a predominantly unknown aetiology (Alzheimer’s (AD) and Parkinson’s disease (PD)) [13]. Mapping disease progression from early through to terminal stages is challenging enough in humans, let alone in animals. More is known about end-stage disease from post-mortem analysis, but the fact that many conditions are asymptomatic initially adds a significant hurdle to studying early stages of disease onset.
Despite advances in genetic sampling, humans have a long lifespan compared with most other species and research still struggles to define the genetic determinants at each stage of disease. The contribution of different genes at developing stages of human life influences disease phenotypes and therefore any predictive model would need to capture phenotypes at different time points [14]. As such, phenotypic diversity is very difficult to model in preclinical studies. For example, AD and PD models are still hugely limited in this respect [15], [16]. For inherited neuropathies, most diseases are length dependent [14]. A human lower limb nerve could be up to 1m in length; not something that could be studied in typical animal models.
An example of complexities in preclinical modelling is shown with Eli Lilly’s solanezumab, a humanized monoclonal antibody that binds to, and promotes clearance of, soluble Aβ plaques in AD [17]. Indeed, transgenic PD-APP mouse models of AD showed a dose-dependent reduction of Aβ deposition [18]. Despite this, in three Phase III trials the drug did not meet its primary endpoints. Patient screening criteria were shifted during the progression of these trials; for example, by EXPEDITION 3 patients were screened for having established Aβ plaques in efforts to improve outcomes [19], [20]. However, the results from the PD-APP mouse models arguably only demonstrated evidence of prevention of plaque formation (mice <9 months old) and were not evidence of activity against established plaques (mice aged 18-21 months) [21]. Retrospectively, the company conceded that a failure to impact patients with established plaques was expected based on animal data. Why then did it proceed with EXPEDITION 3 at all? Of course, various other reasons potentially contributed to the Phase III failures of solanezumab as well, including lack of imaging in earlier phases and the subsequent scepticism about the role of Aβ plaques. This, controversially, has recently led to the accelerated approval of Aducanumab (Aduhelm), based on the surrogate endpoint of Aβ plaques reduction. It remains to be seen whether the required post-approval trial will show a clear translation of this endpoint into clinical benefit.
The importance of reproducibility in CNS animal testing was shown with a large-scale study performed by the ALS Therapy Development Institute (TDI) in Cambridge, Massachusetts. It tested more than 100 drugs in an established mouse model of Amyotrophic lateral sclerosis (ALS) in which many of the drugs had been previously reported to show efficacy [22]. Despite this, none of the drugs tested were found to be beneficial in their experiments (Figure 1).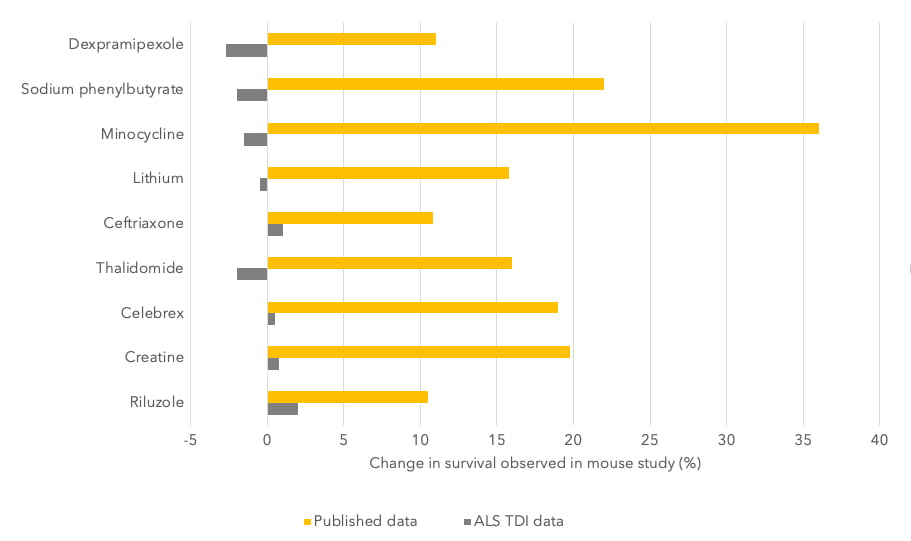
Figure 1 – Results of animal tests by the Amyotrophic Lateral Sclerosis Therapy Development Institute (ALS TDI) versus those published. Image taken from reference [22].
Eight of these compounds ultimately failed in clinical trials, which together involved thousands of people. Even Riluzole, an approved treatment for ALS, showed no survival benefit in the preclinical studies performed by the institute verses those published. This highlights the importance of testing across different laboratories to ensure the validity of results.
Autoimmune and inflammatory
As treatments move away from global immunosuppression and towards more targeted therapies, it is more important than ever to use relevant and representative animal models in preclinical testing of immunomodulatory therapies.
Most autoimmune diseases develop due to a combination of genetic and environmental factors. For example, molecular mimicry is a concept by which the impact of environmental factors might be potentiated, and autoimmune processes accelerated. Critical inflammatory factors may constitute targets for immune intervention and thus, to evaluate therapies, such as whether a specific blockade of a critical cytokine, the corresponding inflammatory factor or cell population should also play a crucial role in the pathogenesis of the disease in animals. Additionally, cardiovascular complications often run alongside autoimmune disorders [23]. Models fail to capture the interplay between autoimmunity and cardiac complications owed to accelerated atherosclerosis, increased systemic inflammation and anti-heart auto-reactivity, which also directly affect cardiac cells and tissues.
Unfortunately, differences between the human and the murine immune systems complicates disease modelling in animals, and as such, models often represent only a certain aspect of disease. Certainly, acute inflammatory and autoimmune disorders can translate well from preclinical to clinical testing. Chronic autoimmune disorders are much harder to capture in animals e.g., multiple sclerosis and lupus. The lack of accuracy in autoimmune models, e.g., autoimmune hepatitis, has significantly hindered the development of innovative therapies [24]. However, there is a huge ongoing focus to humanize animal models in this field, as discussed later.
An example of failing to predict the differences between immune systems of different species is shown with theralizumab (TGN1412), a superagonist CD28-specific monoclonal antibody initially developed by TeGenero for use in autoimmune conditions [25]. Despite a lack of toxicity in preclinical testing at much higher doses, in the initial trial all six volunteers experienced severe adverse effects such as fever, headache, hypotension, and lymphopenia, and ultimately multi-organ-failure. These severe adverse events were attributed to the induction of cytokine release syndrome predominantly originating from CD4+ memory T-cells, which produced high levels of TNF-α, IL-2 and IFN-γ. Several failures in preclinical development have been proposed, firstly being a lack of CD28 expression on the CD4+ effector memory T-cells of species used for pre-clinical safety testing of TGN1412 could not predict a ‘cytokine storm’ in humans [26]. Secondly, that the preclinical test did not include a test for allergy. This was important because CD28 is also expressed by the cells responsible for allergy and the fact that the adverse reactions were immediate, relates to the release of preformed cytokines in granules of allergy-mediating immune cells [27].
Perspectives
Preclinical animal models will never be completely representative of human disease, and no amount of optimisation will overcome inherent species to species variability. In the wild, mice die from natural causes like predation or parasites, this is often reflected in their natural immune system. Representation is worsened by strain differences through inbreeding, with knock-in and knock-out modifications further artificializing animal models. Ultimately, the further away you move from fundamental, natural physiology, the more artificial the results become.
The impact of poor preclinical planning is huge and is wasteful of animal life, resources and time, and risks lives of clinical trial participants. In cases of slow-developing neurological conditions, patients may have only one shot at preventing progression. Inadequate preclinical hypotheses and conclusions can be devasting and misdirect funding and time that could be spent elsewhere.
The misuse or misplacement of animal modelling is likely more prolific in the early research space, where funds are limited, and PoC demonstration in in vivo models are an often-unspoken requirement for investors or industry partners. Changing these ‘requirements’ and allowing researchers freedom to demonstrate reasoning for not conducting animal experiments is critical. We need to move away from preclinical modelling being a tick-box exercise and a barrier for engagement, funding, or licensing.
Researchers should invest time to understand the limitations of preclinical models. Eton et al proposed a decision tree for preclinical model evaluation and selection, highlighting when it is time to give up and seek alternatives [13]. Engagement with regulators to have upfront discussions before embarking on preclinical experiments that do not provide relevant answers or predictions of human experience can be invaluable, and often worth the cost. We have seen first-hand the benefits of these early discussions and the support from regulators to adapt standard requirements for complex, and often rare, diseases.
We of course need to continually improve methods of studying human phenotypes to provide baseline requirements more accurately for animal modelling [14]. For example, studying extreme disease phenotypes can help identify relevant disease modifying genes. Using molecular imaging to map disease progression over time in humans can help validate phenotypic developments in animal disease models [28]. For now, a rationalised balance of multiple preclinical experiments can provide separate insights into one or more aspects of cancer behaviour. Collaboration is key, consortia like MODEL-AD, which is optimising new mouse models able to predict later-onset disease will make its models freely available. Unfortunately, there are still cases where companies block academic use of certain strains with patented DNA [15].
Even with appropriate disease models, adequate powering of studies, rigorous outcome measures and the need for study replication in independent laboratories is critical to obtaining closer predictions. A clear statistical analysis plan is key not just in clinical testing, but also preclinical. Accounting for possible bias in experiments is also key e.g., selection bias can be overcome with randomization and detection bias can be addressed with blinding [29]. A study of abstracts presented at the Society for Academic Emergency Medicine (SAEM) annual meetings showed that from 290 animal models used, 194 were not randomized and 259 were not blinded; these studies were shown to have a 3.4- and 3.2-fold increased likelihood of claiming statistical significance in findings [30], [31]. Another analysis by the UK-based National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs) found that only 59% of 271 randomly chosen articles stated the hypothesis or objective of the animal study, and the number and characteristics of the animals used. Most papers did not report using randomisation (87%) or blinding (86%) to reduce bias in animal selection and outcome assessment.
The use of positive controls is another crucial experimental consideration and is something we commonly see as absent in preclinical data. These controls are fundamental in validating experimental findings and are an important basis for understanding any true treatment benefit [32]. Only 70% of the publications that used statistical methods fully described them and presented the results with a measure of precision or variability [33]. Collaborating with experienced biostatisticians or preclinical developers (even within the same institute) can provide impactful advice and save time, money, and animal life in generating meaningful preclinical insights.
It is also important to encourage publication of ‘bad’ results. The validity of published literature is a significant driver in all areas of life sciences - publication bias is well known in clinical trial data, but it is also important in early-stage research. An analysis of animal stroke studies showed that efficacy can be overestimated by ca. 30% when negative results are unpublished [34]. Better transparency in experimental findings could prevent researchers from moving into disproven avenues of research and instead focus programs into other areas.
Future outlook on animal testing
There are many new methodologies and technologies used and in development to both optimise (Refine or Reduce) and to Replace typical animal models. We discuss some examples below.
Refinement and Reduction
Refinement includes ensuring the animals are provided with housing that allows the expression of species-specific behaviours, using appropriate anaesthesia and analgesia to minimise pain, and training animals to cooperate with procedures to minimise any distress [2]. Evidence suggests that pain and suffering can alter an animal’s behaviour, physiology, and immunology. Such changes can lead to variation in experimental results that impairs both the reliability and repeatability of studies.
Endpoints
Selecting appropriate endpoints can significantly contribute to reduced pain in animals. For example, selecting endpoints that occur before observable suffering or clinical signs of a condition. Examples might include the use of imaging technologies to assess internal tumour burden or the use of study-specific biomarkers in serum. Identifying earlier scientific endpoints may allow more refined humane endpoints to be implemented, thereby minimising pain and suffering in the models used. A greater use of molecular imaging can also avoid animal sacrifice and permits repetitive longitudinal studies on the same animal which becomes its own control [35].
Clinical protocols, for example in tumour imaging, are more standardized, achievable on a variety of systems, and typically limit scan durations for patient comfort and to accommodate scanner schedules. In contrast, preclinical imaging programs are commonly project- and machine-specific, limiting their dissemination and broad employment. Preclinical scanners, for example, must image at much higher resolution than clinical scanners, requiring different hardware solutions. Small animal imaging techniques include µPET, µSPECT, µMRI and µCT.
An ongoing collaborative ‘co-clinical’ sarcoma study is measuring human response to pembrolizumab, surgery, and radiation alongside response to treatment in a genetic mouse model using imaging [36]. The clinical study is sponsored by Sarcoma Alliance for Research through Collaboration in collaboration with Stand Up To Cancer and MSD, with data analysed against preclinical experiments performed at Duke University. The study aims to create a blueprint for µMRI and µCT as a resource for public dissemination of preclinical imaging data, protocols, and results. The study demonstrated similar imaging in the clinical and preclinical studies and addressed a principal challenge in assessing tumour burden in the lungs with µCT due to the effect of respiratory motion on images. The group employed respiratory gating techniques to mitigate the effects of motion by limiting acquisition to defined periods in the breathing cycle. Tissue barriers, such as diaphragm, lung wall, and tumour boundaries, were visibly clearer in gated images. Tumour volume measurements from gated images more accurately matched post-mortem standards.
Humanizing models
Humanized animal models aim to mimic components of human disease. Humanization is particularly important when studying immunotherapies in animal models given the vast difference in immune systems. For example, in models of rheumatoid arthritis (RA) two main strategies have been studied: 1) introduction of human transgenes e.g., human leukocyte antigen molecules of T cell receptors, and 2) generation of mouse/human chimera by transferring human cells or tissues into immunodeficient mice [37]. A review of current methodologies by Schinnerling et al compares traditional models of RA with those incorporating humanization methodology and shows that a combination of transgenic expression of RA risk alleles and the engraftment of RA patient-derived immune cells and/or RA synovial tissue seems to be a promising strategy to avoid GvHD and establish chronic autoimmune responses. Similarly, Christen et al discusses this in the context of autoimmune hepatitis and the need to extend research into humanization strategies to allow expanded drug development [24].
Work conducted at Yale to produce representative animal models of myelodysplastic syndromes (MDS) used a patient-derived xenotransplantation model in cytokine-humanized immunodeficient “MISTRG” mice [38]. These mice expressed five humanized growth factors shown to be permissive for human haematopoiesis and support robust reconstitution of human lymphoid and myelomonocytic cellular systems. Of course, this relies on access to human patient samples.
CrownBio has developed HuGEMM, a platform for immunotherapy evaluation using immunocompetent chimeric mouse models engineered to express humanized drug targets, such as genes encoding for immune checkpoint proteins, which can be directly used to evaluate therapeutics [39]. Another is MuPrime, which consists of tumour homograft models that have been engineered to incorporate human disease pathways. MuPrime models featuring oncogenic drivers, for example, can be used to evaluate responses to targeted therapies and/or combination immunotherapies.
Use of large animal models
Interspecies differences between humans and animals are unavoidable in preclinical testing; above discussions centre on humanizing animal conditions, however, another option is to use larger animals in preclinical studies. There are certain biological upsides to using smaller animal models e.g., reduced genetic variation, short generation intervals, high fecundity, and ease of maintenance and handling, translating into both cost and time savings [40]. However, larger animals offer a more accurate anatomical scale and capture, for example, the faster rate of metabolism which impacts the timeline for disease development. Larger animals can also be studied for longer periods of time; swine for example can survive up to 10 years.
Swine models offer large animal benefits at a lower cost than non-human primates. They have been used extensively in toxicology testing; however, research has also explored their use in modelling disease and treatment. Schachtschneider et al highlights the lack of a genotypically, anatomically, and physiologically relevant large animal model in oncology and has developed transgenic porcine models (Oncopig) as a next-generation large animal platform for a multitude of cancers [40]. It also models comorbidities like obesity and liver cirrhosis alongside primary cancer indications.
There are significant ethical considerations for studying human disease in non-human primates. Despite this, work is ongoing to develop such models for neurological conditions like AD and PD which are severely limited in mouse models. The European Union is sponsoring a consortium of research organizations called IMPRiND that is aiming to develop a standardized macaque model of AD by injecting them with brain tissue from humans, which leads the animals to develop plaques and tau tangles, as well as cognitive impairment [15]. This approach has already produced a primate model of PD. At RIKEN, Saido et al is creating a marmoset model of AD by using CRISPR to insert mutations into fertilized eggs [41]. Marmosets can develop both Aβ plaques and tau tangles more quickly than macaques.
Model selection frameworks
Various frameworks have been developed to try to quantify the representation of human disease in animal models and to optimise the selection process. Sams-Dodd and Denayer et al proposed a scoring system which allows the selection of one or more animal models to represent human disease most accurately [42], [43]. The framework scores five different categories from 1 to 4: species, disease simulation, face validity (number of symptoms represented), complexity and predictability. A more recent framework, FIMD (Framework to Identify Models of Disease) by Ferreira et al studies different categories, scoring from 1 to 12.5 the epidemiology, symptomatology and natural history, genetics, biochemistry, aetiology, histology, pharmacology, and endpoints [44].
The authors apply the framework to preclinical models of Duchenne muscular dystrophy (mouse versus dog) and show quantifiably the benefits of the dog model. However, the authors highlight a current limitation in applying the model for diseases with poorly understood aetiology. Until a complete comprehension of human disease and the technical ability to represent this in animals is defined, frameworks like those mentioned have limited applicability. However, in certain disease scenarios they could have merit.
Experimental design
Guidelines have been developed to assist researchers, particularly earlier stagers who have not been exposed to animal testing, in developing rigid testing criteria. The European Union (EU) Directive 2010/63 refers to guidelines for education, training and competence, and for the housing, care and use of research animals [45]. The NC3R also offers extensive guidance and training resources across all areas of animal testing. The organization awards funding for research, innovation, and early career training to a wide range of institutions to accelerate the development and uptake of 3Rs approaches.
Building on the EU Directive, Smith et al developed PREPARE (Planning Research and Experimental Procedures on Animals: Recommendations for Excellence) guidelines aimed to improve quality, reproducibility and translatability of data [46]. PREPARE is a 15-point checklist for scientists to address before starting preclinical testing and includes not only their own methods but also considers the wider legal and ethical aspects as well as the quality and division of labour with partnering centres.
Reporting data
Systemic reviews of animal studies can help promote translation of preclinical studies into the clinic and can avoid unintended waste for other researchers. The ARRIVE (Animals in Research: Reporting In Vivo Experiments) guidelines were the first harmonised guidelines to establish reporting standards in animal research [44], [47]. They consisted of a checklist of 20 items describing the minimum information that all scientific publications reporting research using animals should include, such as the number and specific characteristics of animals used (including species, strain, sex, and genetic background); details of housing and husbandry; and the experimental, statistical, and analytical methods (including details of methods used to reduce bias such as randomisation and blinding).
The GRADE (Grading and Recommendations Assessment, Development and Evaluation) guidelines have been applied in analysing clinical data and were more recently explored for analysing the quality of published animal research [48]. The approach uses five factors for rating down the quality of evidence (risk of bias, indirectness, inconsistency, imprecision, and publication bias) and three for rating up (magnitude of effect, dose-response gradient, plausible confounding). The outcome is a grading of evidence from high, moderate, low, or very low. These types of criteria and assessment guidelines can assist researchers not only in reporting high quality results, but also during planning processes for experimental design. As mentioned earlier, encouraging the publication of all data is essential, including ‘bad’ results.
Replacement
In certain cases, human disease is not, and will likely never be, represented in animal models. The human brain is far more complex than its mouse counterpart; for example, neurons in the human cortex arise from a cell type that is not present in relevant levels in rodents (outer radial glia) [49]. There is also a significant difference in metabolism; humans developing far more slowly. Humans are also not inbred, which is important given the impact of genetic diversity on disease onset and progression.
NC3R highlights two Replacement routes:
- Partial replacement - the use of some animals that, based on current scientific thinking, are not considered capable of experiencing suffering. This includes invertebrates and immature forms of vertebrates. Partial replacement also includes the use of primary cells and tissues taken from animals killed solely for this purpose (not having been used in a scientific procedure that causes suffering).
- Full replacement - avoids the use of any research animals. It includes the use of human volunteers, cells and tissues, mathematical and computer models, and established cell lines.
Artificial intelligence
AI is unlikely to directly replace animal modelling; however, it can be used as a data mining tool to refute or validate hypotheses before animal models are considered and is often essential in combination with microfluidics models. As quoted by Juan Carlos Marvizon at UCLA “computers can do amazing things, but they cannot guess information that they do not have” [50].
Many of the current collaborations in this field focus on earlier drug discovery e.g., molecular dynamics and molecular docking methods. There has been significant partnership between big pharma and AI companies on the drug development front. Pfizer is using IBM Watson, a system that uses machine learning, to power its search for immuno-oncology drugs [51]. Sanofi is working with UK start-up Exscientia to hunt for metabolic-disease therapies, and Genentech is using an AI system from GNS Healthcare in Cambridge, Massachusetts, to help drive the company’s search for cancer treatments. Most sizeable biopharma players have similar collaborations or internal programs.
As an example of data mining, a group at Johns Hopkins used machine learning to analyze published toxicity data, compiling a database of 300,000 chemicals with associated biological data [52]. This database allowed the group to predict the toxicity of a given chemical based on similarity of structure before conducting animal studies. The analysis found that several chemicals within the database had been excessively tested in repetitive toxicology models which caused unnecessary loss of animal life. One could argue that researchers must repeat experiments themselves, but perhaps this is a pattern to break and more trust should be given to already published data as suggested in previous sections.
Interestingly, Verisim Life is developing digital animal simulations to potentially circumvent the need for animal testing - or at least to provide early predictive read outs and reduce animal testing requirements as part of screening.
2D cells and tissues
The commercialisation of induced pluripotent stem cells (iPSC) was first based on the landmark discovery in 2006, in Japan, by Yamanaka whereby the addition of a small number of defined transcription factors reprogrammed or ‘induced’ a somatic adult cell into an embryonic stem-cell like state (as an iPSC) capable of cell division into other cell types. Even now, companies in this space pay fees and/or royalties to iPSC Academia Japan for use of its iPSC reprogramming licenses.
The primary focus for 2D iPSC assays has been their use in toxicology testing with the displacement of primary human hepatocytes, for example. The first commercially available iPSC cell product was launched in 2009 by Reprocell as a human iPSC-derived cardiomyocyte product (ReproCario). However, companies and researchers are also beginning to focus on using iPSCs for disease modelling which could replace, or at a minimum reduce, animal use in drug screening. This has been accelerated with the inclusion of gene-editing to the iPSC workflow. For example, Reprocell has a proprietary CRISPR-SNIPER technology and is supplying disease models in the retinal, neurodegenerative, and metabolic areas. Definigen also has a proprietary workflow and is developing disease models in the liver space, including inherited diseases, toxic injury and infectious disease.
iPSC-disease modelling has also allowed generation of postmitotic neurons and glial cells, otherwise only available from sensory nerve biopsies and post-mortem samples [53]. Patient-specific iPSC-derived neural cells recapitulate the genotype and phenotypes of disease and have proven to be successful for studying several neurodegenerative diseases including frontotemporal dementia, AD, PD, Huntington disease, spinal muscular atrophy, ALS, Duchenne muscular dystrophy, schizophrenia, and autism spectrum disorders and recently in Charcot-Marie-Tooth disease. Nevertheless, at the mechanistic level, comparison with animal models still show discrepancies, primarily owed to the variability in methodology.
Culturing methods have continued to improve, and are now yielding 3D cell cultures, spheroids, and organoids that more accurately reflect human tissues.
3D organotypic tissue slice cultures
Organotypic cultures consist of sectioned tumour tissue into thin slices, mounted onto porous membranes for mechanical support and incubated in a controlled condition [54]. They retain histological and 3D structure with inter- and extracellular interactions, cell matrix components, and intact metabolic capacity. This approach has been successfully used to gain insights into tumour biology and as a preclinical model for drug discovery in many different cancers e.g., lung, prostate, colon, gastric, pancreatic and breast, among others.
Widespread use of these cultures in personalized oncology has lagged due to the absence of standardized methods for comparison between samples. There has also been a lack of evidence demonstrating that these cultures reflect clinical characteristics of human cancers. However, a recent study by Kenerson et al reported a standardised method to assess and compare human cancer growth ex vivo across a wide spectrum of GI tumour samples, capturing the state of tumour behaviour and heterogeneity [55].
Mitra Biotech’s CANscript platform uses this principle to generate a patient-specific phenotypic assay to measure multiple parameters and determine tumour response to selected treatments [56]. The measurements are converted into a single score, known as an ‘M-Score’, that predicts a patient’s clinical response to the tested therapies. These assays are of course limited by access to adequate patient samples.
3D stem cell organoids
Organoids have been used to study infectious diseases, genetic disorders, and cancers through the genetic engineering of human stem cells, as well as directly from patient biopsy samples [49]. These are the most likely models to serve as viable alternatives to animal testing. Organoid models generally use either pluripotent (PSC) or adult stem cells (AdSC). The latter are isolated directly from patients but are limited by access and supply. iPSCs have been positioned to replace the use of AdSCs in 3D organoids and organ-on-a-chip models for use in studying human disease.
During formation differentiated iPSCs aggregate to first form an organ bud, and later organoids, that mimic the mature organ structure, including multiple cell types and the interactions between them [49]. The 3D arrangements are typically created by the addition of biocompatible materials such as hydrogels. Clearly iPSCs can overcome the limitations of supply, and hepatic 3D organoids derived from iPSCs show advanced hepatic differentiation when compared to their 2D iPSC counterparts and are scalable for clinical and high-throughput applications [57].
iPSC organoids have been developed for study in a variety of disease areas including the brain, liver, esophagus, stomach, colon, intestine, and lung. Some of the larger commercial players include Hubrecht Organoid Technology, STEMCELL Technologies, Cellesce, DefiniGEN, Qgel and OcellO, Organovo, and InSphero.
In neurology, the creation and study of brain organoids, sometimes termed ‘mini-brains’, is a somewhat contentious area of research. Various groups have successfully used these models to study AD pathologies [15]. For example, Lin et al used iPSCs carrying the APOE ε4 variant, which were shown to secrete a stickier Aβ, and which encouraged aggregation of Aβ and tau [58]. Using CRISPR to edit the cells so that the risk-conferring variant ε4 was replaced with the risk-neutral variant ε3 led to a reduction in signs of the condition, including increased Aβ production, in neurons, immune cells and even organoids. Park et al created a 3D triculture model using neurons, astrocytes, and microglia in a 3D microfluidic platform which mirrored microglial recruitment, neurotoxic activities such as axonal cleavage, and NO release damaging AD neurons and astrocytes [59]. AxoSim has a commercially available brain organoid model (BrainSim) developed initially at Johns Hopkins; it also has a nerve model, NerveSim, claiming to have high levels of Schwann cell myelination and human-relevant electrical and structural metrics.
Despite these successes, studies have also shown that organoids do not yet recapitulate distinct cellular subtype identities and appropriate progenitor maturation [60]. Although the molecular signatures of cortical areas emerge in organoid neurons, they are not spatially segregated. Organoids also ectopically activate cellular stress pathways, which impairs cell-type specification. Additionally, iPSC organoids across the board currently show high variability and require expensive and sophisticated manufacturing protocols.
3D organ-on-a-chip
Organ-on-a-chip has been evolving rapidly over the last decade and goes one step further than organoids, offering multicellular systems with microfluidics, thus imitating organ function and environment [61], [62]. These chips range from devices the size of a USB pen drive to larger systems that reflect multiple linked organs within the footprint of a 96-well plate [63]. All platforms have three characteristics: 1) the 3D nature and arrangements of the tissues on the platforms; 2) the presence and integration of multiple cell types to reflect a more physiological balance of cells; 3) the presence of biomechanical forces relevant to the tissue being modelled.
In oncology for example, microfluidics techniques enable researchers to capture the inherent shear fluid pressures that are found in the native microenvironment of the tumour, a feature that can ‘turn on’ inherent drug resistance mechanisms, and dynamically influence heterogeneity of spheroidal cell clusters [64]. Just like the ongoing use of organoids in personalized medicine, organ-on-a-chip offers the same opportunity. A breast cancer study by Shirure et al demonstrated a platform able to facilitate the culture, growth, and treatment of tumor cell lines, as well as patient-derived tumor organoids and to visualize angiogenesis, intravasation, proliferation, and migration at high spatiotemporal resolution, and quantifies microenvironmental constraints such as distance, flow, and concentration, that allow a tumor to communicate with the arterial end of the capillary [65].
An example showing the power of iPSCs coupled with genome editing technologies, investigated Barth syndrome where stem cell-derived cardiac tissues from patient donors were created and modelled on ‘muscular thin films’, which replicated the disordered sarcomeric organization and weak contraction properties of the disease [66]. Genome editing to ‘correct’ the faulty TAZ gene in the iPSC-derived cells identified mitochondrial abnormalities that were unknown.
Most studied organ chip models include those replicating the heart, liver, and lung. Commercial players include Emulate, a Wyss Institute start-up commercializing the Institute’s organ chip technology and automated instruments. Others include MIMETAS, Kirkstall, and Hurel Corp.
Tissue chips offer promise in modelling multiple organs and tissues from individual donors of both healthy and diseased disposition and investigating the responses of these tissues to the environment and therapeutics. However, each tissue requires specific supply of relevant nutrients and growth factors, therefore, linked tissue systems have a key challenge in providing universal cell culture medium or ‘blood mimetic’ [63]. Despite this, many organs have been explored with these systems, including liver, lung, heart, kidney, intestine in various indication areas [62]. US governmental funding from the Defence Advanced Research Project Agency was allocated to create a 10-organ ‘body-on-a-chip’ system [63]. A subsequent publication showed how a 10-organ chip combined with quantitative systems pharmacology computational approaches could model the distribution of in vitro pharmacokinetics and endogenously produced molecules [67].
Researchers from the Hebrew University recently used their tissue-on-a-chip technology as a complete replacement for animal testing, showing that diabetes drug empagliflozin could be used alongside cisplatin to reduce the occurrence of nephrotoxicity [68]. The group has supposedly submitted this research to the FDA for review, but the outcome is not yet public [69].
3D bioprinting
Bioprinting has been around for a long time and can alleviate some hurdles by precise and controlled layer-by-layer assembly of biomaterials in a desired 3D pattern [70]. Researchers are finding innovative ways to combine organoid technologies with 3D bioprinting (synergistic engineering). both organoids, which self-organize into three dimensions, and bioprinted tissues, can be seeded or printed in multi-well plates with inclusion of biomechanical forces, creating platforms with multi-tissue components that may be amenable to larger-scale commercial production [63].
A team at the Wyss Institute has developed a bioprinting method that generates vascularized tissues composed of living human cells that are ten-fold thicker than previously engineered tissues and that can sustain their architecture and function. The method uses a silicone mold to house tissue on a chip, where the mold has a grid of larger vascular channels containing endothelial cells into which a self-supporting ‘bioink’ containing MSCs is printed. After this, a liquid composition of fibroblasts and ECM components fills any open regions adding the connective tissue component.
Building representative organs with this method certainly has remaining challenges. Including the development of additional bioinks to ensure organ function post-printing and creating more cell-friendly methods, where shear stress during printing can impede cell growth and can even change expression profiles [70]. One of the key focusses in this area is advancing the technology for regenerative medicine purposes.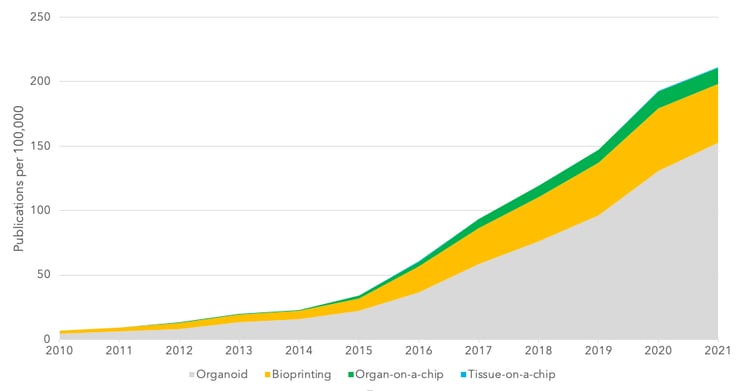
Figure 2 – Key word search for emerging technologies. Graphs shows the number of articles with positive key word search per 100,000 articles in the complete database.
Conclusions
We have discussed some techniques and emerging technologies aimed at reducing or replacing animal modelling. Organoid models and use of iPSCs certainly seem to be the most advanced option and there is increasing research using these models (Figure 2). In fact, analysts project a CAGR of 22.5% between 2020 and 2027, with a market value forecast in 2027 of $2b in this sector [71]. However, there are several key limitations in widespread adoption of these models. High cost is a significant factor and something that forces companies into using cheaper animal models. Another limitation is on the available range of models and the reliable production of models to commercial scale.
The preclinical process of Big Pharma is to discontinue projects quickly to increase translational success. The quickest and cheapest way to do this is parallel testing products in multiple animal models of the same disease; this is not a process that could be easily adapted to non-animal methods. If larger companies, with relatively limitless resources are unwilling to move to more expensive non-animal models there is an even lower probability that earlier-stage researchers will make this move.
Animal testing is not only a pivotal facet of drug development, in most countries it is a legal requirement that must be completed before clinical trials can commence. It is effectively seen as a check-box exercise.
It is not only the developers and regulators that would need to be convinced; importantly the clinical investigators must relate to the data presented in Investigator Brochures (IB) and have confidence in the translation of results into their trials. If the preclinical data presented to investigators as part of the IB is deemed unconvincing or insufficient, the trial in question may not be a favoured choice for that investigator to lead. Patient bias is a real issue, and clinical researchers may favour ‘ideal patients’ e.g., young, fit and otherwise healthy, for early clinical trials for products they feel have the most promising and reliable preclinical data. Therefore, it is important to move away from unreliable preclinical animal testing, but equally important to present data from reliable assays that investigators are familiar with and can relate to.
Methods are limited by what we can know in the human system: no single system completely recapitulates a fully functional and integrated human tissue, let alone an organ. Rather, systems are designed to model key aspects or characteristic by mimicking the morphological and functional phenotype of interest. Likely, newer methods will run alongside animal models until they could be optimised to allow replacement.
References
[1] W. Russell and R. Burch, “The Principles of Humane Experimental Technique by W.M.S. Russell and R.L. Burch,” John Hopkins Bloomberg School of Public Health, 1959. .
[2] “https://www.nc3rs.org.uk.” .
[3] E. Törnqvist, A. Annas, B. Granath, E. Jalkesten, I. Cotgreave, and M. Öberg, “Strategic focus on 3R principles reveals major reductions in the use of animals in pharmaceutical toxicity testing,” PLoS One, 2014, doi: 10.1371/journal.pone.0101638.
[4] A. D. Hingorani et al., “Improving the odds of drug development success through human genomics: modelling study,” Sci. Rep., 2019, doi: 10.1038/s41598-019-54849-w.
[5] D. Cook et al., “Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework,” Nature Reviews Drug Discovery. 2014, doi: 10.1038/nrd4309.
[6] C. Belzung and M. Lemoine, “Criteria of validity for animal models of psychiatric disorders: focus on anxiety disorders and depression,” Biol. Mood Anxiety Disord., 2011, doi: 10.1186/2045-5380-1-9.
[7] C. R. Ireson, M. S. Alavijeh, A. M. Palmer, E. R. Fowler, and H. J. Jones, “The role of mouse tumour models in the discovery and development of anticancer drugs,” British Journal of Cancer. 2019, doi: 10.1038/s41416-019-0495-5.
[8] C. H. Wong, K. W. Siah, and A. W. Lo, “Estimation of clinical trial success rates and related parameters,” Biostatistics, 2019, doi: 10.1093/biostatistics/kxx069.
[9] E. Borcoman, A. Nandikolla, G. Long, S. Goel, and C. Le Tourneau, “Patterns of Response and Progression to Immunotherapy,” Am. Soc. Clin. Oncol. Educ. B., 2018, doi: 10.1200/edbk_200643.
[10] E. Pan, D. Bogumil, V. Cortessis, S. Yu, and J. Nieva, “A Systematic Review of the Efficacy of Preclinical Models of Lung Cancer Drugs,” Front. Oncol., vol. 10, p. 591, 2020.
[11] M. V. Guerin, V. Finisguerra, B. J. Van den Eynde, N. Bercovici, and A. Trautmann, “Preclinical murine tumor models: A structural and functional perspective,” eLife. 2020, doi: 10.7554/eLife.50740.
[12] E. Guerin, S. Man, P. Xu, and R. S. Kerbel, “A model of postsurgical advanced metastatic breast cancer more accurately replicates the clinical efficacy of antiangiogenic drugs,” Cancer Res., 2013, doi: 10.1158/0008-5472.CAN-12-4183.
[13] S. L. Eaton and T. M. Wishart, “Bridging the gap: large animal models in neurodegenerative research,” Mammalian Genome. 2017, doi: 10.1007/s00335-017-9687-6.
[14] M. Reilly and A. Rossor, “Humans: the ultimate animal models,” J. Neurol. Neurosurg. Psychiatry, vol. 91, pp. 1132–1136, 2020.
[15] A. King, “The search for better animal models of Alzheimer’s disease,” Nature. 2018, doi: 10.1038/d41586-018-05722-9.
[16] S. Salari and M. Bagheri, “In vivo, in vitro and pharmacologic models of Parkinson’s disease,” Physiological Research. 2019, doi: 10.33549/physiolres.933895.
[17] A. Sun and L. Z. Benet, “Late-Stage Failures of Monoclonal Antibody Drugs: A Retrospective Case Study Analysis,” Pharmacology. 2020, doi: 10.1159/000505379.
[18] R. B. DeMattos, K. R. Bales, D. J. Cummins, J. C. Dodart, S. M. Paul, and D. M. Holtzman, “Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease,” Proc. Natl. Acad. Sci. U. S. A., 2001, doi: 10.1073/pnas.151261398.
[19] R. S. Doody et al., “Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease,” N. Engl. J. Med., 2014, doi: 10.1056/nejmoa1312889.
[20] L. S. Honig et al., “Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease,” N. Engl. J. Med., 2018, doi: 10.1056/nejmoa1705971.
[21] R. B. DeMattos et al., “A Plaque-Specific Antibody Clears Existing β-amyloid Plaques in Alzheimer’s Disease Mice,” Neuron, 2012, doi: 10.1016/j.neuron.2012.10.029.
[22] S. Perrin, “Preclinical research: Make mouse studies work,” Nature, 2014, doi: 10.1038/507423a.
[23] C. Sanghera, L. M. Wong, M. Panahi, A. Sintou, M. Hasham, and S. Sattler, “Cardiac phenotype in mouse models of systemic autoimmunity,” DMM Dis. Model. Mech., 2019, doi: 10.1242/dmm.036947.
[24] U. Christen, “Animal models of autoimmune hepatitis,” Biochimica et Biophysica Acta - Molecular Basis of Disease. 2019, doi: 10.1016/j.bbadis.2018.05.017.
[25] M. D. E. Goodyear, “Further lessons from the TGN1412 tragedy,” British Medical Journal. 2006, doi: 10.1136/bmj.38929.647662.80.
[26] D. Eastwood et al., “Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4 + effector memory T-cells,” Br. J. Pharmacol., 2010, doi: 10.1111/j.1476-5381.2010.00922.x.
[27] J. H. Weis, “Allergy test might have avoided drug-trial disaster [3],” Nature. 2006, doi: 10.1038/441150c.
[28] Y. Waerzeggers, P. Monfared, T. Viel, A. Winkeler, and A. H. Jacobs, “Mouse models in neurological disorders: Applications of non-invasive imaging,” Biochimica et Biophysica Acta - Molecular Basis of Disease. 2010, doi: 10.1016/j.bbadis.2010.04.009.
[29] W. Huang, N. Percie du sert, J. Vollert, and A. Rice, “General Principles of Preclinical Study Design,” Good Res. Pract. Non-clinical Pharmacol. Biomed., pp. 55–69, 2019.
[30] V. Bebarta, D. Luyten, and K. Heard, “Emergency medicine animal research: Does use of randomization and blinding affect the results?,” Acad. Emerg. Med., 2003, doi: 10.1111/j.1553-2712.2003.tb00056.x.
[31] J. P. A. Ioannidis, “Extrapolating from animals to humans,” Science Translational Medicine. 2012, doi: 10.1126/scitranslmed.3004631.
[32] P. Moser, “Out of Control? Managing Baseline Variability in Experimental Studies with Control Groups,” Good Res. Pract. Non-clinical Pharmacol. Biomed., pp. 101–117, 2019.
[33] C. Kilkenny et al., “Survey of the Quality of Experimental Design, Statistical Analysis and Reporting of Research Using Animals,” PLoS One, 2009, doi: 10.1371/journal.pone.0007824.
[34] E. S. Sena, H. Bart van der Worp, P. M. W. Bath, D. W. Howells, and M. R. Macleod, “Publication bias in reports of animal stroke studies leads to major overstatement of efficacy,” PLoS Biol., 2010, doi: 10.1371/journal.pbio.1000344.
[35] V. Cuccurullo, G. Di Stasio, M. Schilliro, and L. Mansi, “Small-Animal Molecular Imaging for Preclinical Cancer Research: .μPET and μ.SPECT,” Curr Radiopharm, vol. 9, no. 2, pp. 102–113, 2016.
[36] S. J. Blocker et al., “Bridging the translational gap: Implementation of multimodal small animal imaging strategies for tumor burden assessment in a co-clinical trial,” PLoS One, 2019, doi: 10.1371/journal.pone.0207555.
[37] K. Schinnerling, C. Rosas, L. Soto, R. Thomas, and J. C. Aguillón, “Humanized mouse models of rheumatoid arthritis for studies on immunopathogenesis and preclinical testing of cell-based therapies,” Frontiers in Immunology. 2019, doi: 10.3389/fimmu.2019.00203.
[38] Y. Song et al., “A highly efficient and faithful MDS patient-derived xenotransplantation model for pre-clinical studies,” Nat. Commun., 2019, doi: 10.1038/s41467-018-08166-x.
[39] M. Labant, “Animal Models Evolve to Satisfy Emerging Needs,” www.genengnews.com, 2020. .
[40] K. M. Schachtschneider et al., “The oncopig cancer model: An innovative large animal translational oncology platform,” Front. Oncol., 2017, doi: 10.3389/fonc.2017.00190.
[41] K. Sato et al., “A non-human primate model of familial Alzheimer’s disease,” bioRxiv, 2020.
[42] F. Sams-Dodd, “Strategies to optimize the validity of disease models in the drug discovery process,” Drug Discovery Today. 2006, doi: 10.1016/j.drudis.2006.02.005.
[43] T. Denayer, T. Stöhrn, and M. Van Roy, “Animal models in translational medicine: Validation and prediction,” New Horizons Transl. Med., 2014, doi: 10.1016/j.nhtm.2014.08.001.
[44] G. S. Ferreira et al., “A standardised framework to identify optimal animal models for efficacy assessment in drug development,” PLoS One, 2019, doi: 10.1371/journal.pone.0218014.
[45] Official Journal of the European Union, “Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes,” 2010.
[46] A. J. Smith, R. E. Clutton, E. Lilley, K. E. A. Hansen, and T. Brattelid, “PREPARE: guidelines for planning animal research and testing,” Lab. Anim., 2018, doi: 10.1177/0023677217724823.
[47] C. Kilkenny, W. J. Browne, I. C. Cuthill, M. Emerson, and D. G. Altman, “Improving bioscience research reporting: The arrive guidelines for reporting animal research,” PLoS Biol., 2010, doi: 10.1371/journal.pbio.1000412.
[48] D. Wei et al., “The use of GRADE approach in systematic reviews of animal studies,” Journal of Evidence-Based Medicine. 2016, doi: 10.1111/jebm.12198.
[49] J. Kim, B. K. Koo, and J. A. Knoblich, “Human organoids: model systems for human biology and medicine,” Nature Reviews Molecular Cell Biology. 2020, doi: 10.1038/s41580-020-0259-3.
[50] J. C. Marvizon, “Computer models are not replacing animal research, and probably never will,” www.speakingofresearch.com, 2020. .
[51] N. Fleming, “How artificial intelligence is changing drug discovery,” Nature, 2018, doi: 10.1038/d41586-018-05267-x.
[52] R. Van Noorden, “Software beats animal tests at predicting toxicity of chemicals,” Nature. 2018, doi: 10.1038/d41586-018-05664-2.
[53] M. Juneja, J. Burns, M. A. Saporta, and V. Timmerman, “Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development,” J. Neurol. Neurosurg. Psychiatry, 2019, doi: 10.1136/jnnp-2018-318834.
[54] A. Salas et al., “Organotypic culture as a research and preclinical model to study uterine leiomyomas,” Sci. Rep., 2020, doi: 10.1038/s41598-020-62158-w.
[55] H. L. Kenerson et al., “Tumor slice culture as a biologic surrogate of human cancer,” Ann. Transl. Med., 2020, doi: 10.21037/atm.2019.12.88.
[56] B. Majumder et al., “Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity,” Nat. Commun., 2015, doi: 10.1038/ncomms7169.
[57] T. Takebe et al., “Massive and Reproducible Production of Liver Buds Entirely from Human Pluripotent Stem Cells,” Cell Rep., 2017, doi: 10.1016/j.celrep.2017.11.005.
[58] Y. T. Lin et al., “APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types,” Neuron, 2018, doi: 10.1016/j.neuron.2018.05.008.
[59] J. Park et al., “A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease,” Nat. Neurosci., 2018, doi: 10.1038/s41593-018-0175-4.
[60] A. Bhaduri et al., “Cell stress in cortical organoids impairs molecular subtype specification,” Nature, 2020, doi: 10.1038/s41586-020-1962-0.
[61] M. Dhandapani and A. Goldman, “Preclinical Cancer Models and Biomarkers for Drug Development: New Technologies and Emerging Tools,” J. Mol. Biomark. Diagn., 2017, doi: 10.4172/2155-9929.1000356.
[62] Q. Wu et al., “Organ-on-a-chip: Recent breakthroughs and future prospects,” BioMedical Engineering Online. 2020, doi: 10.1186/s12938-020-0752-0.
[63] L. A. Low, C. Mummery, B. R. Berridge, C. P. Austin, and D. A. Tagle, “Organs-on-chips: into the next decade,” Nature Reviews Drug Discovery. 2020, doi: 10.1038/s41573-020-0079-3.
[64] G. C. Kocal et al., “Dynamic Microenvironment Induces Phenotypic Plasticity of Esophageal Cancer Cells under Flow,” Sci. Rep., 2016, doi: 10.1038/srep38221.
[65] V. S. Shirure et al., “Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids,” Lab Chip, 2018, doi: 10.1039/c8lc00596f.
[66] G. Wang et al., “Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies,” Nat. Med., 2014, doi: 10.1038/nm.3545.
[67] C. D. Edington et al., “Interconnected Microphysiological Systems for Quantitative Biology and Pharmacology Studies,” Sci. Rep., 2018, doi: 10.1038/s41598-018-22749-0.
[68] A. Cohen et al., “Mechanism and reversal of drug-induced nephrotoxicity on a chip,” Sci. Transl. Med., 2021, doi: 10.1126/scitranslmed.abd6299.
[69] https://www.iflscience.com/, “First Drug Developed Using No Animal Testing Submitted For FDA Approval,” 2021. .
[70] M. Dey and I. T. Ozbolat, “3D bioprinting of cells, tissues and organs,” Scientific Reports. 2020, doi: 10.1038/s41598-020-70086-y.
[71] Grand View Research, “Organoids And Spheroids Market Size, Share & Trends Report Organoids And Spheroids Market Size, Share & Trends Analysis 2020-2027,” 2020.