Intro:
For decades, conventional drug strategy has relied on the ability of small molecules to either inhibit or alter protein function by targeting their active sites. While this was instrumental in modern medicine, the clinical potential of small molecules is restricted by the druggability of the protein target. Within the roughly 20,000 expressed human proteins, 2500 are thought to be disease related, with fewer than 30% of these being targeted by currently approved drugs (1).
These limitations are inspiring researchers to move past a ‘protein-only’ mindset in drug development and instead, explore other biomolecules as potential therapeutic targets. One avenue that has gained considerable momentum in recent years is that of RNA-targeted therapeutics. Interest in this field has been intensified due to the ‘RNA revolution’, which has created a fundamental shift in how researchers perceive ‘the central dogma’ - the process by which the instructions in DNA are converted into a functional product (2). Thanks to advances in novel transcriptomic technologies, RNA is now emerging as a more diverse class with important roles in genetic regulatory control and a whole host of cellular activities.
Recent advancements in our understanding of ubiquitous functional RNA molecules in human cells have further added to the rising interest in RNA targeting. The Encyclopaedia of DNA Elements (ENCODE) project uncovered that approximately 70–90% of the genome is transcribed to RNA, while only 3% of those transcripts code for protein, indicating that most of the genome yields noncoding RNAs (ncRNAs) (3,4). This study allowed for mapping of previously unidentified long noncoding RNAs (lncRNAs), short microRNAs (miRNAs), and other ncRNAs in a variety of cell types (2).
Since then, many of these newly discovered ncRNAs have been found to be disease associated, both in cancer and nontumorigenic diseases (2). Antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs) and more recently, small activating RNAs (saRNAs), have added to the identification of therapeutically manipulable target RNAs.
However, small molecule drugs remain the preferred entity for disease treatment, owing to their more favorable pharmacokinetic and pharmacological properties. Moreover, their ability to bind to and modulate secondary and tertiary RNA structures, has made these drugs complementary tools to sequence-recognizing ASOs. While the field of RNA-targeted small molecules remains well in its infancy, success of discovery efforts will open unprecedented opportunities for therapeutically modulating cellular processes that have previously been considered 'undruggable'.
1. RNA CLASSES
While the traditional view of RNA casts the molecule as a conduit for transferring information encoded in the nuclear genome to the cytoplasmic translational machinery, RNA is now emerging as a more diverse class. Aside from messenger RNAs (mRNAs) encoding proteins, transfer RNAs (tRNAs) which carry amino acids, and ribosomal RNA (rRNA) involved in translating them, understanding continues to grow of the repertoire of ncRNA classes and their diverse functions.
ncRNAs are generated from the larger part of the genome that does not encode proteins but produces noncoding transcripts that regulate gene expression and protein function. While there are several ncRNAs, the two major classes of ncRNA that will be discussed are the miRNAs and the more recently identified lncRNAs. Dysregulation of both types of transcripts are associated with every cancer and affects all major cancer hallmarks (5). Additionally, they have been linked to complex biological processes such as immune cell development and function, immune disorders, neural development, and neurological diseases. As a result, therapeutic targeting of such naturally occurring ncRNAs therefore represents a very promising approach for the treatment of various diseases.
miRNAs
miRNAs are small single-stranded non-coding RNA molecules. miRNA is synthesized as primary miRNAs that are first processed by the enzyme Drosha and subsequently by Dicer in the cytosol, to form a single-stranded miRNA product around 17–22 nucleotides long (2,6).
The mode of action of miRNAs involves recognition of the 3ʹ-untranslated region (3ʹ-UTR) of target mRNAs through sequence complementarity and recruitment of nucleases to repress mRNA expression. The 2-8 nucleotides at the 5ʹ-end of miRNAs is the seed region, which is conserved among miRNA family and directs target recognition. High complementarity causes mRNA degradation while partial complementarity prevents target mRNAs from being translated. One miRNA can have multiple targets; therefore, dysregulation of a single miRNA can result in extensive changes in gene expression. Thus, the miRNA-mRNA networks inside cells are extremely complicated, and it is also difficult to find a developmental or physiological process without the influence of miRNAs.
lncRNAs
lncRNAs are RNAs with >200 nucleotides that lack coding function. The ENCODE project identified nearly 10,000 lncRNAs, many of which have been found to have numerous cellular functions including the regulation of chromatin architecture, transcriptional regulation, inhibition, or enhancement of protein activity and guiding epigenetic modification complexes, to name a few (3).
lncRNAs are also understood to be dysregulated in disease, a key example being lncRNA metastasis associated lung adenocarcinoma transcript 1 (MALAT1) which is important in normal pre-mRNA splicing (7). However, MALAT1 overexpression appears to influence numerous cancer processes.
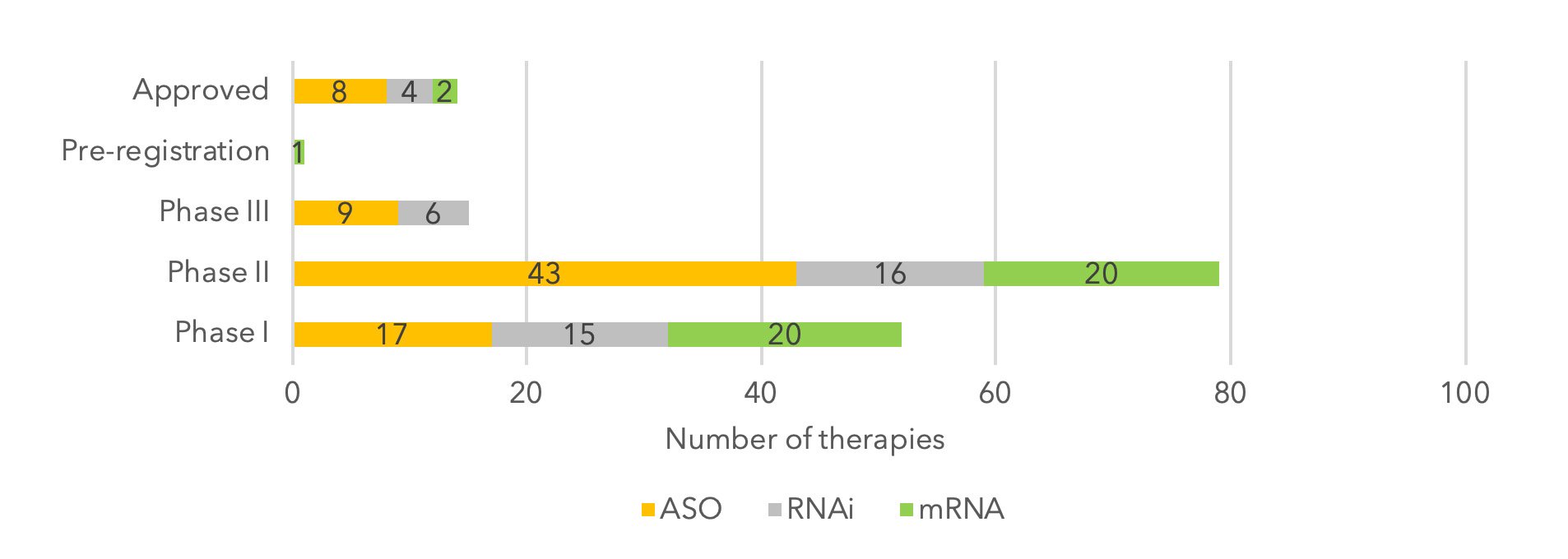
Figure 1: RNA therapy clinical pipeline by stage of development.
2. RNA THERAPEUTICS LANDSCAPE
The major advances in knowledge surrounding the diversity, structural and functional information related to RNAs has put these biomolecules in the limelight as promising drug targets for a multitude of diseases including bacterial/viral infections, neurodegenerative diseases, and cancer. However, the fast-growing interest in RNA targeting has been met with several challenges, including the lack of diversity in building blocks, and increased structural flexibility of RNA.
Nevertheless, strong proof of principle for RNA-targeted drugs has been provided by clinical trials investigating the use of ASO and RNA interference (RNAi), which is mediated by double-stranded RNA molecules called short interfering RNAs (siRNAs) (8).
Broadly speaking, RNAi operates sequence specifically and post-transcriptionally by activating ribonucleases which, along with other enzymes and complexes, degrade the RNA after the original RNA target has been cut into smaller pieces. In contrast, ASOs bind to their target nucleic acid via Watson-Crick base pairing, and inhibit or alter gene expression via steric hindrance, splicing alterations, or initiation of target degradation, to name a few. Through a range of mechanisms, this typically silences gene expression by reducing transcription of the mRNA. In some cases, where certain genes are repressed, slight nuances in the target design of the ASOs can also facilitate a positive correction, either through modifying splicing to increase production of full-length functional proteins or increasing expression by designing an RNA sequence complementary to the promoter region of a gene (8).
In recent years, the drug discovery platforms built by pioneers such as Alnylam and Ionis have been able to create viable new oligonucleotide-based candidates at pace, progressing through clinical development with high degrees of success. Currently, there are eight ASOs approved for the treatment of five separate indications and four RNAi products on the market, with Amondys 45 being the most recent to enter the market (9).
While ASOs have remained the most common and developed technology type, earlier development stages are filled with RNAi and mRNA. In addition, oligonucleotides that target regulatory gene regions to upregulate expression, also known as small activating RNAs (saRNAs), have attracted interest from big pharma companies in recent years.
MiNA Therapeutics is the frontrunner in saRNA development and has recently partnered with Eli Lilly, AstraZeneca and Servier. Other biotechnology companies including Stoke Therapeutics, Ravtigen and CAMP4 Therapeutics are also expanding their portfolios of saRNA candidates designed to enhance gene expression.
Overall, the recent spate of approvals for RNA-targeted therapeutics in the last 12 months, together with the global awakening to the potential of mRNA vaccines amidst the COVID-19 pandemic, has accelerated the creation of new pipeline assets. Today, there are at least 690 RNA therapies currently in development (from preclinical to pre-registration stage), with 20 of these in Phase III clinical trials (9). This is a 50% increase from 2020, highlighting the accelerated interest in RNA-based therapeutics.
3. PITFALLS OF RNA THERAPEUTICS
The translation of oligonucleotide-based therapies into clinical use has been hampered by their enhanced susceptibility to ribonuclease (RNase) degradation, their inability to cross the cell membrane due to their strong negative charge, and their activation of the innate immune system (11). While chemical modifications to and formulations of RNA molecules have dramatically increased their stability, including the use of lipid nanoparticles which function by encapsulating large amounts of RNA and protecting them against RNase degradation and renal clearance, achieving effective delivery to many tissues outside of the liver remains a significant translational challenge (12).
A major challenge associated with the systemic delivery of these drugs to the central nervous system (CNS) is due to their inability to traverse the blood–brain barrier (BBB) (11). This was exemplified by the recent failure of Roche’s tominersen and a pair of ASOs from Wave Life Sciences in their Phase III and Phase I/II trials, respectively (13). Both products were aimed at lowering levels of the resulting mutant form of huntingtin in HD. The patient post-mortems following these trials showed that a combination of insufficient gene silencing and inadequate penetration of brain structures contributed to their failures, prompting commentators to question the longevity of ASOs as a technology (13).
However, despite challenges, 13 oligonucleotide-based drugs were approved by the FDA in the last two decades with more being approved each year.
4. EMERGENCE OF RNA-TARGETED SMALL MOLECULES
The problematic clinical delivery of oligonucleotide-based therapies has led to renewed interest in small molecules targeting the more complex RNA structures and/or the dynamic landscape of RNA. Unlike DNA, RNA molecules have been shown to have stable tertiary, and in some cases, quaternary structures. The development of new biophysical and structural techniques aimed at obtaining a more complete picture of the folding of long transcripts has unearthed short- and long-range interactions that create unique, higher-order folding structures as well as the presence of biologically relevant conformational landscapes (14).
These insights have provided scientists with a new opportunity for selective RNA targeting with small molecules that could lead to the activation or inhibition of RNA functions or alter gene expression and cellular processes toward the control of disease (15). In addition to their higher specificity for RNA compared to sequence-based methods, the extensive chemical and biophysical tunability of small molecules, as well as their ‘drug-like’ physicochemical and pharmacokinetic properties, make them appealing alternatives (16).
However, despite the vast amount of potential RNA targets in the transcriptome, targeting RNA with small molecules has remained on the periphery of drug development for some time. This is due to the fundamental challenges associated with RNA targeting including the dynamicity of RNA and the lack of specificity for RNA target engagement in a cellular environment where 85% of the total RNA is ribosomal (17). Additionally, in contrast to protein drug targets, which comprise a total of 22 proteinogenic amino acids, RNAs are comprised of only four primary nucleotide monomeric units as building blocks. With each containing a nucleobase, ribose, and phosphate group, nucleotide monomers are substantially more complex than individual amino acids (15).
Nevertheless, there are several approved antibiotic drug classes dating back several decades, including natural and semisynthetic, tetracyclines and macrolides, as well as synthetic oxazolidinones such as linezolid and tedizolid, whose mechanisms of action involve selective binding to rRNAs within the 30S or 50S subunits to interfere with protein synthesis for the control of infections. The success of these antibiotics in the treatment of various bacterial infections, has provided sound support for the notion that RNA is a legitimate target of small molecules (15).
Beyond antibiotics targeting rRNAs, several other approaches have been pursued to target RNA with small molecules, one being riboswitches – regulatory elements commonly found in the 5′-untranslated region (UTR) of mRNAs that exert their regulatory control over the transcript in a cis-fashion by directly binding a small molecule ligand (18). Binding of small molecules induces a conformational change that allows for gene transcription or translation to be regulated in bacterial systems.
One important example of small molecules targeting riboswitches is Merck’s ribocil which was discovered serendipitously through phenotypic screening of small molecules interfering with the bacterial riboflavin biosynthetic pathway. Ribocil acts to inhibit the flavin mononucleotide (FMN) riboswitch and disrupt translation of the downstream mRNA (1). Although ribocil selectively binds the FMN riboswitch, the rapid development of bacterial resistance in various species has ultimately precluded this drug from progressing as a clinical candidate in humans (1). Nevertheless, the development of ribocil has been valuable in proving that phenotypic screens, which have not traditionally considered RNA as a potential target, can in fact identify small molecules that bind RNA.
RNA splicing modifiers
Interest in RNA targeted small molecules in treating human diseases has increased with the success of several drug candidates involved in modifying RNA splicing – an intricate process that cells use to remove unwanted regions of their RNA code (introns) and stitch the desired coding regions, called exons, together. Mutations that lead to altered regulation of splicing are the direct cause of many human diseases, including many forms of cancer, myotonic dystrophy, and spinal muscular atrophy (SMA).
To date, there have been few small molecules targeting RNA splicing; Table 1 summarizes the current clinical pipeline. PTC Therapeutics has developed a small molecule, Translarna (ataluren), that reads through a stop codon in Duchenne's muscular dystrophy (DMD), where 5-10% of DMD patients have a truncated dystrophin gene due to a mutation that creates an internal stop codon. Preclinical studies showed that Translarna allows read-through of nonsense codons in human and mouse cells. The European Medicines Agency granted this therapy conditional approval in 2014 based on a randomized double-blind trial in 174 patients, and recently expanded the label to include children under 5 (19).
|
Company |
Drug |
Named targets |
Likely lead indication |
Status |
|
PTC therapeutics |
Ataluren |
Premature stop codons |
DMD |
Launched (Feb 2014) |
|
Roche/PTC |
Risdiplam |
SMN2 mRNA |
SMA |
Launched (Aug 2020) |
|
Novartis |
Branaplam |
HTT |
Huntington’s disease |
PhIIb |
|
PTC therapeutics |
PTC518 |
HTT |
Huntington’s disease |
PhI |
|
H3 Biomedicine Inc. |
H3B-880 |
SF3b |
CML |
PhI |
|
Eisai |
E7107 |
SF3b |
Cancer |
PhI (suspended) |
Table 1: Clinical pipeline small molecule RNA splicing modifiers (10).
Roche’s risdiplam (Evrysdi) and Novartis’s branaplam (LMI070) are two small molecules that have also recently emerged from phenotypic screens. Both risdiplam and branaplam have partially overlapping mechanism of action; they function by stabilizing the transient double-strand RNA structure formed by the SMN2 pre-mRNA and U1 snRNP complex (20). While risdiplam was recently approved for the treatment of SMA in 2020, Novartis made the decision in July 2021 to discontinue the clinical development of branaplam due to rapid advancements in the SMA treatment landscape, and the fact that this drug would not offer a highly differentiated treatment solution for the SMA community (21).
Nevertheless, Novartis is continuing to advance the investigation of branaplam for HD, a rare, genetic disease with no approved, disease-modifying therapies. In preclinical models, branaplam has been shown to reduce levels of mutant huntingtin protein, and in the SMA program it was also observed to reduce huntingtin messenger RNA in SMA patients. A decrease of huntingtin mRNA is likely to result in the reduction of huntingtin protein levels (HTT), the underlying cause of HD. Novartis has since unveiled plans to start a Phase IIb trial in HD patients in 2021, with the FDA already granting orphan drug designation for branaplam in HD (22).
PTC Therapeutics is also pursuing more RNA splicing modifiers for other disease indications involving 5′ splice sites with imperfect complementarity to the U1 small nuclear RNA (snRNA) component of U1 snRNP. It has recently moved another splice modifier, PTC518, into a Phase I trial in HD (23). This drug is intended to reduce the production of mutant huntingtin protein by promoting the inclusion in huntingtin mRNA of a ‘pseudoexon’ containing a premature stop codon, which is normally spliced out during pre-mRNA processing.
Aside from neurodegenerative diseases, several small molecule splicing modulators have also been evaluated in the clinic for the treatment of solid tumours and leukaemia. One example is Eisai’s E7107 (pladienolide B), the first compound of a new class of anticancer agents targeting the spliceosome (24). E7107 interacts with subunit 1 of SF3b to block the normal splicing of oncogenes. Unfortunately, the development of E7107 was suspended after Phase I clinical trials due to an unacceptable toxicity profile.
H3 Biomedicine is also working on developing novel small molecules that target RNA splicing in cancer. It has recently developed a small molecule modulator of a spliceosome complex SF3b (H3B-8800). This drug offers the benefit of preferentially killing spliceosome-mutant cancer cells whereas other spliceosome inhibitors, such as E7107, show no such preferential targeting (25). H3B-8800 was granted orphan drug status by the FDA in August 2017 and is in clinical trials for the treatment of myelodysplastic syndromes, acute myeloid leukaemia and chronic myelomonocytic leukaemia.
Another biopharmaceutical company pioneering the discovery of a novel small molecules modifying RNA splicing is Skyhawk Therapeutics. This Massachusetts-based start-up has attracted the attention of big pharma, striking several partnerships in recent years including a $74 million partnership with Biogen, a $60 million deal with Celgene, and partnerships with Genentech, Merck & Co., and Takeda Pharmaceutical of undisclosed size (26). Skyhawk is developing small molecules that alter RNA splicing via several distinct strategies including exon inclusion, exon skipping, and exon interference. Their goal is to target historically undruggable proteins, like KRAS, implicated in cancer and prevent the toxic build-up of proteins linked to neurological diseases like Alzheimer’s and Parkinson’s, as well as in inflammatory and infectious diseases.
Overall, the exciting clinical developments that have emerged from previous RNA-splicing modifiers such as Translarna, risdiplam and branaplam highlight the potential to develop small molecule therapies via altering the outcome of alternative RNA splicing events. However, an increased understanding of their complex modes of action, particularly when these compounds exert their effect at RNA-protein interfaces, will be critical for the development of RNA splicing modifiers in the future (27).
miRNAs
In addition to RNA splicing modulators, small molecules directly targeting microRNAs (miRNAs) have also emerged as a promising therapeutic strategy for several disease indications. Understanding of miRNA in disease has greatly advanced due to genome-wide identification of miRNA-mRNA target interactions, the application of RNA sequencing to detect consequences of non-physiological levels of miRNA, and the availability of gene and miRNA expression data from both cell lines and tissue from diseased patients.
Since miRNA has wide-spread regulatory roles, it is not surprising that aberrant expression of miRNAs greatly contributes to human diseases. Indeed, it is now understood that miRNAs play a crucial role in the initiation and development of several human cancers (27). For instance, miR-221 overexpression contributes to hepatocarcinogenesis through targeting DNA damage-inducible transcript 4 (DDIT4) and miR-34 prevents the progression of lung cancer through targeting MET and BCL-2 (27).
miRNAs are not only dysregulated in cancers but can also act as oncogenes or tumor suppressors. Oncogenic miRNAs (oncomiRs) function either by inhibiting tumor suppressor genes or genes responsible for promoting apoptosis or cell differentiation and are normally upregulated in cancer (28).
The corresponding antagomirs, also known as anti-miRNAs, are a class of chemically engineered oligonucleotides that prevent other molecules from binding to a desired site on an mRNA molecule to suppress these oncomiRs resulting in inhibition of tumor growth (28). In contrast, tumor suppressor miRNAs are downregulated in cancers and function by inhibiting oncogenes or genes that hinder apoptosis or cell differentiation.
This widespread involvement of miRNAs across human cancers suggests their utility as new ideal therapeutic targets. In fact, several strategies already exist for modulating dysregulated miRs. These include ASOs; miR sponges which possess complementary binding sites to the miR of interest to “sequester out” its function; and CRISPR/Cas9 based genome editing to modify the genome by delivering Cas9 and guide RNAs to a particular target (28).
However, these strategies are not without their limitations. Like all oligonucleotides, ASOs and miR sponges are not only expensive to develop, but also suffer from poor delivery and decreased in vivo stability (29). In addition, while CRISPR/Cas9 is an extraordinary technology, it is known to cause permanent modifications to the production of miRNA that could induce harmful downstream effects. These challenges are further exacerbated by the ethical implications associated with modifying one’s genome. In this regard, small molecule modulators of miRNA function could be more favourable alternatives (30).
The first small molecule inhibitor of miRNA was reported in 2008 for inhibition of miR-21, which is one of the earliest identified oncomiRs, targeting numerous tumor suppressor genes (31). The mir-21 inhibitor was identified utilizing a luciferase-based assay, which involved transfecting a lentiviral reporter construct containing the complementary sequence of miR-21 downstream of a luciferase gene into HeLa cells, which are known to express high levels of endogenous miR-21 (31).
Since then, several miRNA inhibitors targeting oncomiRs have been identified using high throughput screening or in silico sequence-based design, with several being selected as potent targets for small molecule miRNA inhibitor development (29). Of note, Matthew Disney and his team at Scripps Research identified 68 known drugs that could bind to members of the RNA library, with four binding RNA particularly strongly including dovitinib, a receptor tyrosine kinase (RTK) inhibitor used to treat breast cancer (32). The team subsequently found that dovitinib binds miR-21, which led them to explore ways to skew its action to block miR-21-induced protein production suppression (32). They found that converting dovitinib to a ribonuclease targeting chimera (RIBOTAC), improved the selectivity of dovitinib for the miRNA about 2500-fold. Furthermore, this research group reported that the injection of the RIBOTAC resulted in fewer cancer nodules in mice with experimentally engineering metastasized breast cancer.
Table 2: Notable miRNAs targeted by small molecules in development (10).
|
Company |
Drug |
Named targets |
Likely lead indication |
Status |
|
Sanofi |
Lademirsen (SAR339375) |
miR-21 |
Alport Syndrome |
PhII |
|
Scripps Research |
Dovitinib |
" |
Cancer |
Discovery |
|
Splicos SAS |
ABX-464 |
miR-124 |
Rheumatoid arthritis; Ulcerative colitis |
PhIII |
|
Oncorus Inc |
ONCR-177 |
" |
Cancer |
PhI |
|
Oncorus Inc |
ONCR-148 |
" |
Cancer |
Preclinical |
|
Oncorus Inc |
ONCR-001 |
" |
Cancer |
Discovery |
Not only have these promising findings sparked a renaissance in developing small molecule modulators of miRNAs for cancer therapy, but they have also led to an increased interest in utilizing these drugs to treat inflammatory diseases. Over the past decade, it has become increasingly apparent that miRNAs are key regulators of the immune response and thus represent promising targets for treating inflammatory diseases. Depending on their target mRNAs, miRNAs can either promote or suppress inflammation and therefore are often used by the immune system as rheostats of activation (33). The induction and spread of inflammation are subject to miRNA-mediated positive and negative feedback loops (33). However, while understanding of miRNA biology remains in its infancy, the development of potential miRNA-based therapies is an active field.
Recently, miR-124 was identified as a negative regulator of inflammation. Its downregulation has been reported in diseases such as rheumatoid arthritis (RA) and in patients with pediatric intestinal failure (34). miR-124 overexpression, on the other hand, inhibits intestinal inflammation by reducing the production of IL-6 and TNF-a-converting enzyme (TACE) through the targeting of STAT3 mRNA; a major cytoplasmic signaling molecule downstream of cytokine receptor engagement (35). Children with active ulcerative colitis (UC) have reduced levels of miR-124 and elevated levels of STAT3 in their colon tissues; this combination is likely to induce inflammation and participate in disease pathogenesis (34).
Since miR-124 is a crucial modulator of inflammation and innate immunity, targeting this miRNA may provide therapeutic restitution of physiological pathways lost in inflammatory diseases when targeted with small molecules. Supporting this concept, a recently discovered small quinoline, ABX464 (Abivax), was shown to selectively upregulate miR-124 in human immune cells and has since been shown to strongly attenuate colitis in mice (34). ABX464 was shown to preserve the integrity of the intestinal architecture and villi while simultaneously reducing the influx of inflammatory cells. Concurrently, proinflammatory soluble mediators (i.e. MCP-1, TNF-a, and IL-6) were reduced by in the colon of the ABX464-treated mice (34).
Following these preclinical findings, ABX164 has since been taken to a Phase IIa clinical study in patients with UC. In this randomized study, patients with moderate-severe UC, resistant or intolerant to at least one of immunomodulators, received either ABX464 or a placebo (35). In rectal biopsies of patients treated with ABX464 for 56 days, miR-124 was increased significantly compared with the placebo group and a larger increase in miR-124 was observed in peripheral blood cells (35). This ABX464-driven induction of miR-124 was sustained for at least a year, demonstrating a long-lasting therapeutic effect of this miRNA targeting small molecule.
In addition to the clinical observations in UC patients, ABX464 has also generated promising results in preclinical models of RA. These findings, together with the impressive preclinical and clinical trial data in UC, further emphasize the potential of small molecule drugs targeting miRNAs such as miR-124 in transforming the treatment of inflammatory diseases in the future.
Overall, small molecules targeting miRNAs represent an extremely promising therapeutic avenue not only for cancer, but also for inflammatory diseases and potentially other non-oncology indications. However, the development of this area is embryonic. The screening of compound libraries and discovery of novel miRNA-targeted small molecule drug candidates is a demanding task and therefore effective prediction models will greatly benefit this process. Combination of computational and experimental identifying approaches is the most efficient strategy. Thus, with the development of sophisticated computer-aided and experiment-based screening systems, it is anticipated that small molecule drugs targeting miRNA will soon emerge into the clinic.
lncRNAs
Like proteins, lncRNAs are equipped with multidimensional architecture. While their secondary and tertiary structures are complex, they represent attractive targets awaiting clearer understanding and exploitation. It is envisioned that specific lncRNA structures can be bound or blocked by small molecules and several small molecule compounds identified by library screening have been proven to be effective inhibitors (36). They have been shown to disrupt lncRNA spatial structure or lncRNA–protein interaction.
As previously discussed, the MALAT1 lncRNA affords tremendous drug potential in oncology. There is a highly conserved element for nuclear expression (ENE) at the 3ʹ end of MALAT1 which ensures that it is not degraded and thus is necessary for the physiological function of MALAT1 (36). In several oncology indications, MALAT1 lncRNA is overexpressed which drives cancer proliferation, thus posing the ENE as an attractive target for small molecules (7). The pipeline for lncRNA products is very early. One group led by Amanda Hargrove confirmed that the triple helix structure at the 3ʹ end of MALAT1 could be selectively targeted by small molecules. This group have previously generated derivatives of a known nucleic acid scaffold called furamidine and identified diphenylfuran p8 (DPFp8) as the first small molecule targeted at the triple helix (37).
Another group have further identified two MALAT1 triple helix binders, SM5 and SM16, through a small molecule microarray platform (38). These molecules were found to modulate triple helix dynamics and reduce both MALAT1 transcript abundance in breast cancer organoids. These findings further displayed the development prospects of small lncRNA-targeting molecules.
While these developments are promising, research into small molecule inhibitors of lncRNAs is still in its infancy, so it is unclear whether this technology will outperform oligonucleotide inhibitor technologies. However, compared with the oligonucleotide drugs, small molecule inhibitors have the advantages of more convenient administration modes and lower cost (39). More critically, small molecules target lncRNAs in a structure-specific manner rather than in a sequence-complementary manner and may orchestrate lncRNA functions without changing their expression, which cannot be achieved via RNAi, ASOs, and CRISPR, because small molecules can only disrupt the binding/interaction between lncRNAs and other biomolecules (39). Extensive research into lncRNAs will be necessary to better understand the mechanisms of action of lncRNAs, the binding pockets within lncRNAs, and importantly, discovering molecules that can bind selectively to specific sites with the pocket.
Modulation of RNA modifying enzymes
Another promising approach involves the modulation of RNA modifying enzymes, an area that has remained therapeutically untapped for some time. From a biological perspective, many have questioned the relevance of this field because neither the dynamic nature of RNA epigenetics nor their functional impact was clear early on. However, increasing evidence suggests that these epigenetic marks play a key role in disease from cancer to infection.
While researchers initially identified mRNA modifications in the 1970s, the consequences of those modifications remained elusive. It wasn’t until the realization that N6 methyladenosine (m6A) likely accounts for most internal methylation marks on mRNA and is altered in diverse processes including cell differentiation and viral infection (15), that the field began to take off. In the last decade, researchers have pinpointed the METTL3–METTL14 methyltransferase complex as likely responsible for laying down the m6A marks on most mRNAs (40). This concept was strengthened following the work of Richard Gregory in 2016, who demonstrated that METTL3 is upregulated in certain cancers and selectively promotes the translation of a subset of mRNAs, many of which are known oncogenes (41). Similarly, in the landmark Nature publication in 2017, it was shown that disrupting METTL3 or METTL16 using CRISPR was able to prevent the growth of acute myeloid leukemia (AML) cells both in vitro and in vivo (42).
These findings provided the first evidence to suggest that RNA methylation is a potential driver of AML oncogenesis and thus constitutes a promising therapeutic target.
STORM Therapeutics, Accent Therapeutics and Gotham Therapeutics have each developed small molecule inhibitors of the METTL3–METTL14 complex (40). Twentyeight-Seven Therapeutics and EPICS Therapeutics are two other notable companies in this area; Table 3 summarises small molecules targeting epigenetic markers expecting to reach Phase I over the next few years.
STORM Therapeutics initially discovered its METTL3 inhibitor through high-throughput screening (HTS), biophysical methods and mass spectrometry (43). STORM has previously shown that in a mouse model of AML, oral dosing of its lead compound reduced both splenomegaly and the number of circulating monocytes.
Table 3: Early pipeline of small molecules targeting RNA epigenetics (10,43)
|
Company |
Named targets |
Likely lead indication |
Status |
|
STORM therapeutics |
METTL3; other methyl transferases |
AML |
2021 |
|
Accent Therapeutics |
METTL3, ADAR1 |
AML, NSCLC |
2021; 2022 |
|
Gotham Therapeutics |
METTL3, undisclosed ‘reader’, undisclosed ‘eraser’ |
AML |
2021 |
|
EPICS Therapeutics |
Undisclosed RNA modifying enzymes |
Cancer |
- |
|
Twentyeight-Seven Therapeutics |
Undisclosed RNA modifying enzymes |
Cancer |
-
|
It is now investigating how its candidate fares in other tumour types, including solid tumours, and are aiming to have a clinical lead in Phase I trials in 2021.
Like STORM, Accent Therapeutics has also identified METTL3 inhibitors using a structure-guided approach, and it hopes to have a compound ready for Phase I trials for 2021 (43).
Gotham Therapeutics, with a METTL3 inhibitor in preclinical development, tried an HTS campaign, a fragment-based approach, and a DNA-encoded library screen in parallel to find hits (43). The most promising chemical matter emerged from fragment screens and subsequent fragment evolution. It is aiming for a 2022 entry into the clinic with its METTL3 inhibitor.
5. THE PATH FORWARD FOR RNA-TARGETING SMALL MOLECULES
As the technology has been improved over the past decade, the latest generation of RNA-targeting small molecules are rapidly progressing through clinical development. However, further work is needed to deconstruct the sequence and structure dependencies of small molecule-RNA interactions.
Currently, there is no general method to probe transcriptome-wide binding of a small molecule, and arguably, such an advance would be transformative (15). Nevertheless, the use of HTS methods as described in this paper, have proved instrumental in identifying new biologically active small molecule scaffolds that bind RNA. The continued success of these technologies will provide a wealth of new opportunities for the development of innovative therapeutics that target RNA in the future.
Another important issue to consider in the development of RNA-targeted small molecules is the highly dynamic conformation of RNA. An emerging strategy to exploit the dynamic nature of RNA is by identifying and trapping inactive or non-functional RNA conformations with small molecules thus altering its conformational landscape. Ultimately, this strategy is likely to shift the view that RNA dynamics is an obstacle for selective targeting and instead, a property that can be leveraged towards specific recognition.
|
|
|
|
|
Furthermore, as with any small molecule, potential off-target effects must be evaluated, as many small molecules that bind RNA targets also bind protein targets. Arguably, the more complex challenge will be to assess the downstream biological consequences of a small molecule binding a particular RNA molecule, given the complexities of RNA regulation and RNA biology.
Other avenues for future work in this field include the use of small molecules to selectivity interact with unique RNA structure or sequences of interest, such as protein-binding motifs, (2). In addition, more research is required to better understand to what extent currently approved protein-targeting therapeutics may bind RNA, whether this is as part of their mechanism of action or rather, due to off-target effects. Similarly, natural products remain under-explored for RNA recognition and their often-complex structures may offer favourable selectivity properties or biological activity that may be of important therapeutic use (2). As these drug discovery efforts accelerate, the cumulative learnings will build a new pharmacological lexicon of RNA-interacting small molecules.
Notwithstanding these remaining obstacles to success, RNA-targeted small molecules remain an exciting frontier in drug discovery and development. Targeting RNA with small molecules will allow for indirect targeting of proteins that are currently “undruggable”. Moreover, targeting ncRNA could potentially lead to a wealth of new therapeutic approaches that are currently out of reach for today’s medicine. Addressing these different biological mechanisms RNA-targeting small molecules could access a universe of new targets with novel approaches.
REFERENCES
-
- Warner K, Hajdin C, Weeks K. Principles for targeting RNA with drug-like small molecules. Nature Reviews Drug Discovery. 2018;17(8):547-558.
- Falese J, Donlic A, Hargrove A. Targeting RNA with small molecules: from fundamental principles towards the clinic. Chemical Society Reviews. 2021;50(4):2224-2243.
- An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57-74.
- Hangauer M, Vaughn I, McManus M. Pervasive Transcription of the Human Genome Produces Thousands of Previously Unidentified Long Intergenic Noncoding RNAs. PLoS Genetics. 2013;9(6):e1003569.
- Winkle M, El-Daly S, Fabbri M, Calin G. Noncoding RNA therapeutics — challenges and potential solutions. Nature Reviews Drug Discovery. 2021;20(8):629-651.
- Cech T, Steitz J. The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell. 2014;157(1):77-94.
- Brown J, Bulkley D, Wang J, Valenstein M, Yario T, Steitz T et al. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nature Structural & Molecular Biology. 2014;21(7):633-640.
- Bajan S, Hutvagner G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells. 2020;9(1):137.
- Datamonitor Healthcare | Pharma intelligence [Internet]. Pharmaintelligence.informa.com. 2021 [cited 9 September 2021]. Available from: https://pharmaintelligence.informa.com/products-and-services/data-and-analysis/datamonitor-healthcare
- Cortellis - Clarivate Analytics [Internet]. Cortellis.com. 2021 [cited 9 September 2021]. Available from: https://www.cortellis.com/intelligence/login.do
- Chery J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc Journal. 2016;4(7).
- Roberts T, Langer R, Wood M. Advances in oligonucleotide drug delivery. Nature Reviews Drug Discovery. 2020;19(10):673-694.
- Kwon D. Failure of genetic therapies for Huntington’s devastates community. Nature. 2021;593(7858):180-180.
- Mustoe A, Lama N, Irving P, Olson S, Weeks K. RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proceedings of the National Academy of Sciences. 2019;116(49):24574-24582.
- Yu A, Choi Y, Tu M. RNA Drugs and RNA Targets for Small Molecules: Principles, Progress, and Challenges. Pharmacological Reviews. 2020;72(4):862-898.
- Sztuba-Solinska J, Chavez-Calvillo G, Cline S. Unveiling the druggable RNA targets and small molecule therapeutics. Bioorganic & Medicinal Chemistry. 2019;27(10):2149-2165.
- Hargrove A. Small molecule–RNA targeting: starting with the fundamentals. Chemical Communications. 2020;56(94):14744-14756.
- Garst A, Edwards A, Batey R. Riboswitches: Structures and Mechanisms. Cold Spring Harbor Perspectives in Biology. 2010;3(6):a003533-a003533.
- Haas M, Vlcek V, Balabanov P, Salmonson T, Bakchine S, Markey G et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscular Disorders. 2015;25(1):5-13.
- Sheridan C. First small-molecule drug targeting RNA gains momentum. Nature Biotechnology. 2021;39(1):6-8.
- Novartis Stopping Work on Branaplam as Oral SMA Therapy [Internet]. SMA News Today. 2021 [cited 9 September 2021]. Available from: https://smanewstoday.com/news-posts/2021/07/26/novartis-stopping-development-branaplam-sma-oral-therapy/
- Novartis receives US Food and Drug Administration (FDA) Orphan Drug Designation for branaplam (LMI070) in Huntington’s disease (HD) | Novartis [Internet]. Novartis. 2021 [cited 9 September 2021]. Available from: https://www.novartis.com/news/media-releases/novartis-receives-us-food-and-drug-administration-fda-orphan-drug-designation-branaplam-lmi070-huntington's-disease-hd
- PTC Therapeutics I. PTC Therapeutics Announces that PTC518 Has Entered into a Phase 1 Clinical Trial for the Huntington's Disease Program [Internet]. Prnewswire.com. 2021 [cited 9 September 2021]. Available from: https://www.prnewswire.com/news-releases/ptc-therapeutics-announces-that-ptc518-has-entered-into-a-phase-1-clinical-trial-for-the-huntingtons-disease-program-301174225.html
- Folco E, Coil K, Reed R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes & Development. 2011;25(5):440-444.
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nature Medicine. 2018;24(4):497-504.
- [Internet]. Cen.acs.org. 2021 [cited 9 September 2021]. Available from: https://cen.acs.org/pharmaceuticals/drug-discovery/Roche-partners-Arrakis-small-molecules/98/i14
- Taladriz-Sender A, Campbell E, Burley G. Splice-switching small molecules: A new therapeutic approach to modulate gene expression. Methods. 2019;167:134-142.
- Fan R, Xiao C, Wan X, Cha W, Miao Y, Zhou Y et al. Small molecules with big roles in microRNA chemical biology and microRNA-targeted therapeutics. RNA Biology. 2019;16(6):707-718.
- Wen D, Danquah M, Chaudhary A, Mahato R. Small molecules targeting microRNA for cancer therapy: Promises and obstacles. Journal of Controlled Release. 2015;219:237-247.
- Van Meter E, Onyango J, Teske K. A review of currently identified small molecule modulators of microRNA function. European Journal of Medicinal Chemistry. 2020;188:112008.
- Feng Y, Tsao C. Emerging role of microRNA-21 in cancer. Biomedical Reports. 2016;5(4):395-402.
- Tazi J, Begon-Pescia C, Campos N, Apolit C, Garcel A, Scherrer D. Specific and selective induction of miR-124 in immune cells by the quinoline ABX464: a transformative therapy for inflammatory diseases. Drug Discovery Today. 2021;26(4):1030-1039.
- Xiao Y, Wang J, Lu W, Cao Y, Cai W. Downregulated expression of microRNA-124 in pediatric intestinal failure patients modulates macrophages activation by inhibiting STAT3 and AChE. Cell Death & Disease. 2016;7(12):e2521-e2521.
- Sun Y, Li Q, Gui H, Xu D, Yang Y, Su D et al. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Research. 2013;23(11):1270-1283.
- Vermeire S, Hébuterne X, Napora P, Wisniewska-Jarosinska M, Kiss G, Bourreille A et al. OP21 ABX464 is safe and efficacious in a proof-of-concept study in ulcerative colitis patients. Journal of Crohn's and Colitis. 2019;13(Supplement_1):S014-S015.
- Chen Y, Li Z, Chen X, Zhang S. Long non-coding RNAs: From disease code to drug role. Acta Pharmaceutica Sinica B. 2021;11(2):340-354.
- Donlic A, Morgan B, Xu J, Liu A, Roble C, Hargrove A. Discovery of Small Molecule Ligands for MALAT1 by Tuning an RNA‐Binding Scaffold. Angewandte Chemie. 2018;130(40):13426-13431.
- Abulwerdi F, Xu W, Ageeli A, Yonkunas M, Arun G, Nam H et al. Selective Small-Molecule Targeting of a Triple Helix Encoded by the Long Noncoding RNA, MALAT1. ACS Chemical Biology. 2019;14(2):223-235.
- Chen Y, Li Z, Chen X, Zhang S. Long non-coding RNAs: From disease code to drug role. Acta Pharmaceutica Sinica B. 2021;11(2):340-354.
- Woodcock C, Yu D, Hajian T, Li J, Huang Y, Dai N et al. Human MettL3–MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discovery. 2019;5(1).
- Lin S, Choe J, Du P, Triboulet R, Gregory R. The m 6 A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Molecular Cell. 2016;62(3):335-345.
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson S et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. 2017;552(7683):126-131.
- Cully M. Chemical inhibitors make their RNA epigenetic mark. Nature Reviews Drug Discovery. 2019;18(12):892-894.
Alacrita pharmaceutical product development support
Alacrita's expert teams are ready to support you through every step of the pharmaceutical product development process, leveraging our scientific, clinical and commercial expertise to help you maximize value at each stage. Please click a service below to learn more or contact us.
Product development services we provide: